The actual pathogenesis of CNS involvement in primary Sjögren’s syndrome remains unclear, but autoimmune-mediated demyelination is a proposed pathogenic mechanism.
The differential diagnosis for the neurological manifestations found in Sjögren’s syndrome is extensive and includes infections (e.g., HIV, syphilis, fungal, bacterial and viral), malignancy, sarcoidosis and other demyelinating syndromes, such as MS and idiopathic transverse myelitis. Serum calcium, angiotensin converting enzyme levels and chest X-ray should be obtained, because sarcoidosis can present with optic
neuropathy and myelopathy. CNS lymphoma should be excluded by performing a CSF and serum lymphoma panel. However, it is important to recognize that intravascular CNS lymphoma may prove difficult to exclude.
Treating Acute Episodes
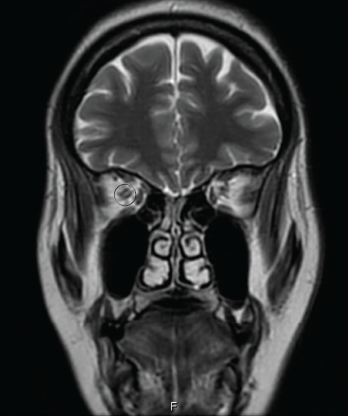
Figure 2: AN ORBITAL MRI
The goals of acute treatment are to suppress the acute inflammatory attack, minimize CNS damage and maintain long-term neurological function. Pulse-dose corticosteroids (e.g., 1,000 mg of IV methylprednisolone) daily for five days, followed by a two- to six-month taper, serve as initial therapy. Plasmapheresis has shown to be effective in patients who are refractory or show minimal response to steroids, making it a good alternative in a patient with coexisting infections.11
Long-Term Management
Azathioprine (AZA) was the first preventive therapy studied in 1998. Subsequent studies confirmed the initial observation of the utility of AZA (3 mg/kg/day) and also indicated the combination of prednisone plus AZA yielded improved outcomes.12
Three studies published from 2009–2014 suggest mycophenolate was more effective in achieving remission in 60–75% of subjects with fewer side effects than azathioprine.13
From 2005–2015, multiple prospective and retrospective rituximab studies indicated sustained remission in up to 83% of NMOSD patients. Further, rituximab was superior to either mycophenolate or azathioprine. Rituximab dosing is either 375 mg/m2 weekly for four doses, or 1,000 mg every two weeks for a total of two doses, followed by scheduled infusions every six months.13
Tocilizumab, an interleukin (IL) 6 inhibitor, has been studied in two pilot studies, because IL-6 is found to be elevated in the blood and spinal fluid of relapsing and active NMOSD patients.14 In the first study, performed in Japan in seven NMOSD patients, tocilizumab (8 mg/kg/month) decreased the relapse rate from 2.9 to 0.4 when added to background immunosuppressive therapy, such as azathioprine or prednisone. IL-6 inhibition also improved neuropathic pain and general fatigue.15 These findings were confirmed in a study performed in Germany in eight NMOSD patients.16
Eculizumab, a C5 inhibitor that blocks the terminal activation of complement and the membrane attack complex, and CD19 monoclonal antibody (MEDI-551), are under investigation to evaluate their ability to prevent NMOSD relapses.12
New treatments have been proposed that target a specific component of disease pathogenesis, including aquaporumab, a recombinant human monoclonal antibody that is composed of an Fc portion binding to AQP4 and thus reduces AQP4-IgG binding, and sivelestat, a neutrophil elastase inhibitor involved in neutrophil migration and phagocytosis.13