An Ancient System for Host Defense
First identified in human serum in the late nineteenth century as a “complement” to Ab in causing bacterial lysis, the complement system emerged more than a billion years ago as what was probably the first humoral immune system. Coral, sponges, sea urchins, and horseshoe crabs have an AP essentially identical to the one humans possess.10 For example, the addition of endotoxin to hemolymph of the horseshoe crab leads to activation of a protease that then triggers both the clotting and complement cascades.11 Crabs don’t want “bad bugs” in their circulatory system any more than humans do!
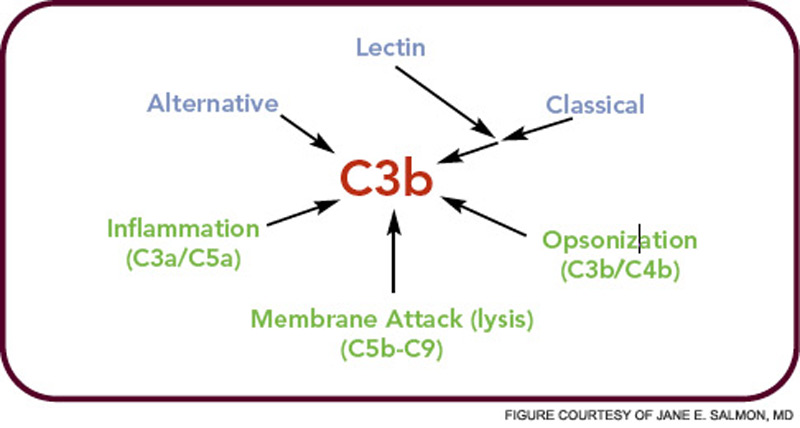
In the crabs, the clotted hemolymph traps the organisms while complement activation coats the bacteria with C3-derived opsonins and liberates the C3a fragment to recruit phagocytes and initiate an inflammatory response. This combination of events is the reason that the complement system is sometimes called “the guardian of the intravascular space.” The AP is truly a simple proteolytic cascade featuring a feedback or amplification loop in which C3 is its central component (see Figure 1, p. 17). The role of the AP in mediating human disease was not appreciated until the past few decades.12,13 Now, it is recognized as an Ab-independent initiator of the acute inflammatory reaction and a potent pathogen recognition system.
The major evolutionary changes that were superimposed on the AP were twofold: one was to facilitate target recognition through Abs, the classical pathway, and lectins, (lectin pathway); the second was the membrane attack (lytic) complex (see Figure 2, p. 17). The AP cascade not only responds to microbes but also to damaged and altered cells and tissues. C3 ticks over at 1% to 2% an hour and, like a sentry, is on guard searching for microbes or damaged self. When it lands on normal self, C3b is inactivated by inhibitors. When it deposits on microbes, unrestricted initial activation is allowed in order to destroy the organism. When it lands on damaged self, a more limited and restricted reaction occurs that is nevertheless sufficient to create an environment for wound repair and to rid the host of the debris.

The Critical Need to Control Complement Activation
Because of the design of the feedback loop coupled with its effector capabilities, tight regulation is critical. To this end, nearly half of the proteins of the complement system act to mitigate its effects in plasma (fluid phase, no target) and on healthy host cells (wrong target).14 However, once cells are injured, limited complement activation is desirable to facilitate tissue repair and to provide for safe removal of debris—the so-called “targeted and restricted complement activation.”15
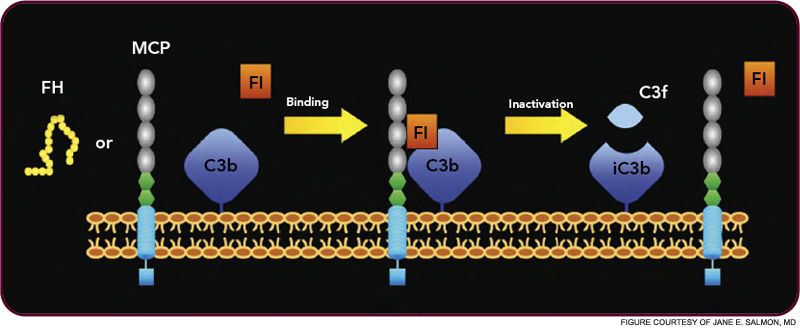
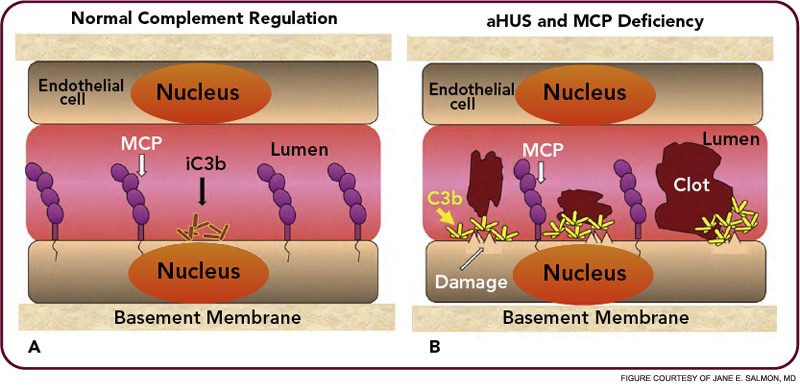
The critical step of C3 cleavage is controlled by regulators that operate through two processes. One mechanism is cofactor activity in which a protein such as the plasma protein factor H (FH) or the nearly ubiquitously expressed membrane cofactor protein (MCP; CD46) binds to C3b (see Figure 3, p. 17). This binding allows a third player, a serine protease of plasma known as factor I, to then degrade C3b. The second process is called decay accelerating activity, in which the catalytic (serine protease) domain of the multiunit C3 convertases is disassociated. These protective mechanisms in large part explain how the complement system separates self from nonself; that is, healthy human tissue is protected, and homeostasis maintained by a constitutively expressed array of regulators on cells and by proteins circulating in plasma. Microbes and damaged cells are not so well shielded. However, a common theme is for opportunistic organisms, especially those that invade the bloodstream, to produce virulence factors that bind the host’s own plasma inhibitors or to synthesize their own CIP mimics.16
Dysfunctions of Regulation Leading to Disease Associations
Cellular wastes, including apoptotic and necrotic cells, need to be efficiently eliminated.17 A simple etiologic explanation for systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) focuses on the consequences of mishandling of DNA/ RNA protein complexes in the former and citrullinated proteins in the latter. Such biological “garbage” can become an immunogen for an adaptive immune response or a nidus for chronic inflammation. Deposition of debris is associated with several of the most common, serious, and lethal illnesses in the developed world: heart attacks and strokes secondary to atherosclerosis (oxidized lipids); dementia in Alzheimer’s disease from misfolded proteins (amyloid); and vision loss in AMD secondary to lipofuscin pigments (drusen). Chronic tophaceous gout would also be in epidemic form, if it were not for our ability to control uric acid levels.
HUS and AMD are diseases representative of two distinct types of complement-mediated injury states. The former is an acute injury that leads to microthrombi. This lesion occurs over a few hours and days and is self-limited. The latter is a situation featuring chronic inflammation over years and decades and is ongoing. Neither condition was thought to have a pathologic relationship to innate immunity, specifically to the complement system. However, whole genome screens identified mutations and polymorphisms in complement regulatory genes as predisposing factors. The basic model arising from these studies is that a reduction in the function of a plasma or membrane inhibitor of the AP permits excessive complement activation on renal endothelium in HUS and on retinal debris in AMD. These associations have much to teach us about the host’s innate immune response to acute injury and to chronic debris deposition.
An Example of Acute Injury State
Atypical hemolytic uremic syndrome (aHUS) is one of a group of conditions termed the thrombomicroangiopathies (TMA) that are associated with endothelial cell injury.2–4 Approximately 50% of patients with aHUS (also called familial, sporadic, nonenteropathic, or Shiga toxin– negative HUS) are haploinsufficient for a CIP. Atypical HUS is characterized by the acute onset of the triad of microangiopathic hemolytic anemia, a low platelet count secondary to consumption, and renal failure caused by microthrombi. “Typical” or the epidemic form of HUS most commonly follows an infection featuring bloody diarrhea by a Shiga toxin– producing Escherichia coli.
Atypical HUS may not have an identifiable trigger, but serious infections in young children are a common precipitating event. Atypical HUS tends to be lethal (25% of patients die acutely) and recurrent (50% of patients develop permanent kidney failure). The penetrance is about 50%, and the mean age at presentation is younger than five years of age. Atypical HUS is a disease of complement dysregulation, commonly featuring a heterozygous mutation in one of three genes: FH, MCP, or factor I.2–4,18 (See Table 1, at left, and Figure 4, p. 18.) Further, gain-of-function mutations for the activating components factor B and C3 have also now been reported in aHUS.19,20 Identification of a mutation is important in disease management owing to differences in mortality, renal survival, and outcome of kidney transplantation.2–4,18 Current treatment is plasma infusion/exchange, but complement inhibitor therapy provides hope for the future.21–23
The clinical, genetic, and immunopathologic understandings gained from studying aHUS illustrate how a powerful recognition and effector system, the AP, reacts to altered self. A response to acute injury must be appropriately balanced. Too much activation or too little regulation promotes undesirable tissue damage. More recently, mutations in these same complement regulatory proteins have been identified in preeclampsia, particularly the HELLP syndrome (hemolysis, elevated liver enzymes, low platelets).24,25 Further, patients with SLE and/or anticardiolipin antibody syndrome who developed preeclampsia had a greater frequency of those mutations.26
Chronic Debris Deposition Disease: The Example of AMD
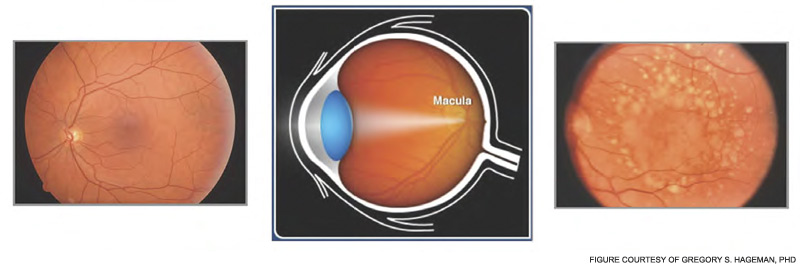
In 2005, four research teams identified a strong genetic link between AMD and a polymorphism in the complement regulator FH.5–8 The lipid/pigment-rich waste known as drusen appears to be a site of an undesirable amount of AP activation (See Figure 5, below).
In 2001, Greg Hageman, PhD, and colleagues demonstrated the presence of C3 fragments, membrane attack complex terminal components, and FH in drusen.9 They proposed a model for AMD pathogenesis based on a prominent role of inflammation. Simply put, according to this model, drusen accumulate, AP is activated, and an excessive inflammatory response damages the retina. Chronic liberation of proinflammatory C3a and C5a anaphylatoxins in the retina are almost certainly deleterious, including their role as a trigger to initiate vascular endothelial growth factor invasion of chorodial vessels (wet AMD) into the macula.27
The polymorphism in FH associated with AMD changes a single amino acid in a region of the protein important for its binding to charged molecules (such as glycosaminoglycans) that are commonly found on cell surfaces and matrices.3 The prevalent hypothesis, for which there is considerable evidence, posits that FH carrying this change (tyrosine in the major allele to a histidine in the minor allele) binds less well to a drusen and therefore results in more AP activation for a given amount of debris. Of great interest, this single amino acid change appears to protect neonates and young children from serious streptococcal infections. Group A beta hemolytic streptococci bind to and coat themselves with FH for protection from complement attack.
Thus, the interesting conclusion of these studies is that the polymorphic FH variant predisposing to AMD is relatively resistant to binding by this pathogen. This feature protects the neonate and young child against such infections. However, this variant FH doesn’t bind as well to drusen and therefore allows for too much retinal inflammation in AMD in later years. The altered activity of the polymorphic variant may not be the primary cause of AMD, which may be degeneration of retinal elements, but it accelerates the process. Thirty to forty percent of the Caucasian population carries the predisposing polymorphic variant.
As this story dramatically illustrates, evolution has naturally focused on getting humans to a reproductive age, not on vision loss in old age. Polymorphisms of this frequency in the human population usually alter function (selecting for or against) and often relate to enhanced survival in early childhood relative to infectious disease.28
Future Therapeutics
Antibody-based therapeutics are a rapidly growing class of drugs. The U.S. Food and Drug Administration approved the first complement-specific therapeutic, the anti-C5 mAb (eculizumab) in March 2007. This agent has proven to be a highly successful therapy for paroxysmal nocturnal hemoglobinuria (PNH).29,30 In PNH, patient red blood cells are deficient in two membrane complement regulators resulting in a life-threatening hemolytic anemia. This mAb blocks the cleavage of C5 to C5a and C5b and halts complement-mediated lysis of the cells. Several case reports also suggest its efficacy in aHUS and clinical trials are in progress.22,23 Other promising drug candidates and complement-targeted therapeutics are on the horizon.
In summary, the innate immune system reacts to injury and cellular debris. The goal of this response is to facilitate tissue repair (i.e., the removal of debris and the prevention of infection). This interaction must be balanced to avoid undesirable consequences such as a microthrombus in aHUS or choroidal neovascularization (wet AMD) in a retina bearing drusen. Importantly, as these studies of disease show, subtle deficiencies in the function of inflammatory regulators may predispose to untoward consequences. Genetic screens have taught us that, in at least two diseases, dysfunctions of the complement system’s AP contribute significantly to pathologies. It is likely that much variation in disease severity in the rheumatic diseases is explained by rather modest alterations in the control of innate immunity.
M. Kathryn Liszewski is a research scientist at the Washington University School of Medicine in Saint Louis. Dr. Atkinson is a professor of medicine and molecular microbiology at Washington University.
References
- Salmon J, de Groot P. Pathogenic role of antiphospholipid antibodies. Lupus. 2008;17:405-411.
- Kavanagh D, Richards A, Atkinson JP. Complement regulatory genes and hemolytic uremic syndromes. Ann Rev Med. 2008;59:293-309.
- Richards A, Kavanagh D, Atkinson JP. Inherited complement regulatory protein deficiency predisposes to human disease in acute injury and chronic inflammatory statesthe examples of vascular damage in atypical hemolytic uremic syndrome and debris accumulation in age-related macular degeneration. Adv Immunol. 2007;96:141-177.
- Fang C, Richards A, Liszewski MK, Kavanagh D, Atkinson JP. Advances in understanding of pathogenesis of aHUS and HELLP. Br J Haematol. 2008;143.
- Edwards AO, Ritter R 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005; 308:421-424.
- Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. 2005;102:7227-7232.
- Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385-389.
- Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005; 308:419-421.
- Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705-732.
- Ariki S, Takahara S, Shibata T, et al. Factor C acts as a lipopolysaccharide-responsive C3 convertase in horseshoe crab complement activation. J Immunol. 2008;181:7994-8001.
- Zhu Y, Thangamani S, Ho B, Ding JL. The ancient origin of the complement system. EMBO J. 2005;24:382-394.
- Thurman JM, Holers VM. The central role of the alternative complement pathway in human disease. J Immunol. 2006;176:1305-1310.
- Holers VM. The spectrum of complement alternative pathway-mediated diseases. Immunol Rev. 2008;223:300-316.
- Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. 2009;9:729-740.
- Riley-Vargas RC, Lanzendorf S, Atkinson JP. Targeted and restricted complement activation on acrosome-reacted spermatozoa. J Clin Invest. 2005;115:1241-1249.
- Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol. 2008;6:132-142.
- Mevorach D, Mascarenhas JO, Gershov D, Elkon KB. Complement-dependent clearance of apoptotic cells by human macrophages. J Exp Med. 1998;188:2313-2320.
- Skerka C, Licht C, Mengel M, et al. Autoimmune forms of thrombotic microangiopathy and membranoproliferative glomerulonephritis: Indications for a disease spectrum and common pathogenic principles. Mol Immunol. 2009;46:2801-2807.
- Fremeaux-Bacchi V, Miller EC, Liszewski MK, et al. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood. 2008; 112:4948-4952.
- Goicoechea de Jorge E, Harris CL, Esparza-Gordillo J, et al. Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci USA. 2007;104:240-245.
- Kavanagh D, Goodship TH, Richards A. Atypical haemolytic uraemic syndrome. Br Med Bull. 2006;77-78:5-22.
- Nurnberger J, Philipp T, Witzke O, et al. Eculizumab for atypical hemolytic-uremic syndrome. N Engl J Med. 2009;360:542-544.
- Gruppo RA, Rother RP. Eculizumab for congenital atypical hemolytic-uremic syndrome. N Engl J Med. 2009;360:544-546.
- Fang CJ, Fremeaux-Bacchi V, Liszewski MK, et al. Membrane cofactor protein mutations in atypical hemolytic uremic syndrome (aHUS), fatal Stx-HUS, C3 glomerulonephritis and the HELLP syndrome. Blood. 2008; 111:624-632.
- Fakhouri F, Jablonski M, Lepercq J, et al. Factor H, membrane cofactor protein and Factor I mutations in patients with HELLP syndrome. Blood. 2008;112:4542-4545.
- Salmon J, Fremeaux-Bacchi V, Liszewski MK, et al. Mutations in complement regulatory proteins predispose to preeclampsia in patients with lupus or antiphospholipid antibodies. ACR/ARHP Annual Scientific Meeting, Vol. In Press. Philadelphia, PA, 2009.
- Gehrs KM, Anderson DH, Johnson LV, Hageman GS. Age-related macular degeneration-emerging pathogenetic and therapeutic concepts. Ann Med. 2006; 38:450-471.
- Alcais A, Abel L, Casanova JL. Human genetics of infectious diseases: between proof of principle and paradigm. J Clin Invest. 2009;119:2506-2514.
- Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol. 2007;25:1256-1264.
- Parker CJ. The pathophysiology of paroxysmal nocturnal hemoglobinuria. Exp Hematol. 2007;35:523-533.