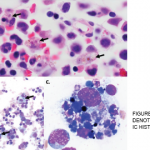
The patient was a 48-year-old woman who saw her primary care physician for a flu-like illness three months prior to admission. Her symptoms initially improved, but recurred one month later; she was treated symptomatically, and again symptoms resolved.
Two months later, she presented to an outside facility’s emergency department with fever to 103ºF, with associated chills, night sweats, headache, fatigue, abdominal pain and arthralgias, which became progressively more severe over one week. She reported having daily fever with headache, fatigue, malaise, myalgias and abdominal pain. There was no recent travel. Her episodes of abdominal discomfort were intermittent and primarily located in the epigastrium and left upper quadrant, without associated nausea, vomiting or diarrhea. Her arthralgias were localized to the bilateral metacarpophalangeal and proximal interphalangeal joints. Review of systems was otherwise negative.
Her past medical history was unremarkable, and she was up to date with routine health maintenance screening, including mammograms and pap smears. The only medication she was taking was acetaminophen for fever and arthralgias. She reported an allergy to penicillin. She had no family history of rheumatic disease. She lived with her husband and had four children. She had quit smoking 15 years earlier, but previously she’d had a 7.5 pack–year history of smoking.
At the outside hospital, she was found to be pancytopenic. This led to an extensive workup that was unrevealing, including tests for EBV, CMV, CMP, leptospira, parvovirus, toxoplasmosis, Babesia, anaplasma, Ehrlichia, Bartonella, influenza and Lyme disease, CT chest/abdomen/pelvis and a bone marrow biopsy. She was transferred to our medical center for additional evaluation.
Upon arrival, she was febrile to 102.2ºF, with a heart rate of 100 beats per minute, blood pressure of 90–100/40–50, and a respiratory rate of 24 with normal oxygen saturation on room air. She was pale in appearance. Her abdomen was soft with tenderness in the left upper quadrant and epigastrium, and the spleen was palpable. Her musculoskeletal exam revealed tenderness at the metacarpophalangeal and proximal interphalangeal joints of both hands, without swelling.
Laboratory studies included a white blood cell count of 2.4 K/µL, hemoglobin of 6.7 g/dL and platelet count of 84 K/µL. Her CRP was elevated to 90 mg/L and her ESR was 33 mm/h. Her ferritin was 543 ng/mL and triglyceride was
325 mg/dL. The remainder of her labs were unremarkable. She had a negative serological workup for ANA, ANCA, RF, anti-CCP and antibodies to RNP, Jo-1, Ro and La. SPEP revealed no abnormalities.