Thanks to research advances in the last decade, we now have a better understanding of the clinically dramatic and often devastating condition known as macrophage activation syndrome (MAS). As a result, says Alexei A. Grom, MD, associate professor of pediatrics at the Cincinnati Children’s Hospital Medical Center, “We are now learning how to watch for the early signs of this condition, treat those patients more aggressively, and prevent the progression of the syndrome to its life-threatening stage.”
In “Macrophage Activation Syndrome: Clinical Presentation and Disease Mechanisms,” presented at the 2009 ACR/ARHP Annual Scientific Meeting in Philadelphia, Dr. Grom outlined the advances in cellular immunology that are contributing to our increasing understanding of MAS. His copresenter, James Verbsky, MD, PhD, assistant professor of pediatrics and microbiology at the Medical College of Wisconsin in Milwaukee, offered two clinical cases that illustrated the diagnostic concepts that he and Dr. Grom discussed.
Features of the Syndrome
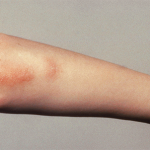
First described in 1979 and defined in 1985, MAS is a potentially fatal complication seen in several rheumatic conditions, including systemic erythematous lupus (SLE) and Kawasaki’s disease.1,2 However, it is most commonly found in systemic juvenile idiopathic arthritis (SJIA). The condition is characterized by excessive activation and proliferation of T cells and well-differentiated non-neoplastic macrophages, Dr. Grom explained. These macrophages exhibit hemophagocytic activity, which leads to a complete destruction of normal hemapoietic elements in the bone marrow. The three cardinal features of MAS are cytopenias, liver dysfunction, and coagulopathy consistent with disseminated intervascular coagulation (DIC). “MAS is also associated with massive systemic inflammation,” he added. “Presentation is often dramatic. Patients develop persistent fevers and impressive generalized lymphadenopathy and hepatosplenomegaly.” Hemorrhagic rash is also characteristic, starting with easy bruising and progressing to severe purpuric lesions and mucosal bleeding. The latter symptoms are found in late-stage disease, along with central nervous system dysfunction. A life-threatening condition, MAS is associated with a 20–30% mortality rate.
What Goes Awry?
“Clinically, MAS is very similar to familial hemophagocytic lymphohistiocytosis [FHLH], a constellation of rare diseases associated with genetic defects that lead to profound depression of cytolytic function of natural killer [NK] cells and cytotoxic CD8 lymphocytes,” Dr. Grom continued. Research has shown that diminished NK cell function in about 40% of FHLH patients is caused by mutations in the perforin gene (PRF1). This research has now yielded a bonus for MAS researchers, Dr. Grom noted, because similar cytolytic defects are seen in patients with SJIA and MAS.
Most cases of MAS are preceded by infection, often a viral infection such as herpes. The pathway to the overexpansion of hemophagocytic macrophages can be understood by first comparing it to the normal immune response, in which NK cells comprise the first line of defense against the pathogen. Antigen-specific cytotoxic CD8 cells become important in subsequent stages of the immune response, killing off the infected cells and secreting multiple cytokines that stimulate macrophages to also kill infected cells. During the normal contraction of the immune response, most of these macrophages undergo apoptosis.
However, as seen in both HLH and MAS, low initial activity of the NK cells, due to mutations in the gene encoding perforin or some other genetic defects affecting the cytolytic pathway, leads to an increased viral load in the early stages of infection. This triggers increased expansion of CD8 cells and sets up a chronic stimulation of macrophages and monocytes, which continue to secrete pro-inflammatory cytokines, including tumor necrosis factor–α, interleukin (IL)-6 and others. Animal studies are now providing support for the central role of cytotoxic CD8 cells secreting interferon gamma. Surplus production of cytokines causes over-activation of the phagocytic macrophages, wreaking havoc on the hemapoietic cells lines and depleting them in the bone marrow.
When you see cytopenias in a patient with systemic [juvenile rheumatoid arthritis], low fibrinogen, and a high ferritin level, this should raise concerns of MAS.
—James Verbsky, MD, PhD
Comparison to SJIA Flares
Many of the symptoms of advanced MAS can resemble flares of rheumatic disease, so diagnosis can be difficult. However, there are important differences that can help clinicians sort out their patients’ symptoms. In both SJIA and MAS, for example, patients have fever, lymphadenopathy, and hepatosplenomegaly. That is where the resemblance ends, noted Drs. Grom and Verbsky. Whereas white blood cell counts, platelets, erythrocyte sedimentation rate, and fibrinogen will typically be elevated in patients having a SJIA flare, these levels are decreased in MAS. SJIA patients’ fevers tend to be quotidian, while those in MAS patients tend to be persistent. The characteristic evanescent rashes seen in SJIA also differ from those in MAS patients, where the rashes are petechial. In addition, ferritin and triglyceride levels will be high in patients with MAS.3
During his presentation, Dr. Verbsky described how his team at the Medical College of Wisconsin used these differences to diagnose two unusual cases. The first was a 12-year-old boy initially admitted to their hospital with the diagnosis of SJIA, due to his fevers of 103–104º, rash, and stiff and swollen joints, as well as other complaints. As is common with these cases, the physicians had started pulse steroid therapy for three days, then switched the patient to oral corticosteroids and methotrexate. Although the boy continued to have intermittent fevers, his rash improved with the corticosteroids, and he was discharged.
Three weeks later, however, the boy was back in the clinic with persistent fevers, breathing problems, and a new petechial rash. His laboratory studies had also changed remarkably, notes Dr. Verbsky. Those results showed that the patient’s white blood cell, hemoglobin, and platelet counts were dropping, a factor inconsistent with a SJIA flare. He also had elevated triglycerides and a high ferritin level (26,000 ng/mL).
These latter results, said Dr. Verbsky, raised a red flag for the clinicians. “When you see cytopenias in a patient with systemic [juvenile rheumatoid arthritis], low fibrinogen, and a high ferritin level, this should raise concerns of MAS,” he noted. The patient was then diagnosed with MAS in association with systemic JIA, and started on the HLH 2004 protocol—comprised of etoposide, dexamethasone, and cyclosporine—and began to improve. Dr. Verbsky called this a “classic presentation of systemic JIA and how it can revert quickly to MAS.”
His second case was a 15-year-old girl whose symptoms met all the clinical criteria for lupus, but who also had unusual features of pancreatitis and hepatitis. She did not respond well to oral prednisone and returned with very high fever as well as elevated triglyceride and ferritin levels. A bone marrow biopsy validated hemophagocytosis, which explained her hepatitis, said Dr. Verbsky. She was diagnosed with MAS in association with systemic lupus erythematosus and admitted to the hospital for aggressive inpatient therapy.
Closing in on Diagnosis
Pediatric rheumatologists are increasingly aware of MAS and this is contributing to its earlier diagnosis, said Dr. Grom. Clinicians should consider the possibility of MAS if their patients have cytopenias, disease that is unresponsive to steroids, unusual organ involvement, and persistent high fevers. Ideally, he said, the diagnosis of MAS should be confirmed by demonstration of hemophagocytic macrophages in the bone marrow, but in some patients these cells accumulate in the lymph nodes, liver, or skin. Sampling errors can also confound bone marrow aspiration results.
Two laboratory tests are useful in diagnosis and monitoring of treatment response, Dr. Grom reported. When T cells and macrophages are overly activated, they shed some of their scavenger receptors, including soluble IL2 receptor and soluble CD163 molecules. Wherever the macrophages accumulate—the bone marrow, lungs, liver, or lymph nodes—they shed these molecules, which can be detected in the peripheral circulation.4 “That’s the reason these two tests may be very helpful in terms of early diagnosis and management of the treatment,” he said. That is also good news in light of evidence that there may be far more cases of occult MAS in juvenile patients than formerly thought.5
The majority of patients with MAS can be successfully treated with a combination of steroids and cyclosporine. For the subgroup of patients diagnosed in the later stages of the syndrome, treatment becomes more problematic. The HLH protocol, with includes steroids, cyclosporine, and etoposide, can be dangerous in children who already have renal or hepatic impairment. It is still unclear whether biologics might be helpful, but beyond case reports of TNF and IL-1 inhibiting agents, there is no clear guidance yet. The key, said Dr. Grom, is to start diagnosing MAS better, at earlier stages. If that occurs, “the overall outcome of this condition will improve.”
Gretchen Henkel is a medical journalist based in California.
References
- Risdall RJ, McKenna RW, Nesbit ME, et al. Virus-associated hemophagocytic syndrome: A benign histiocytic proliferation distinct from malignant histiocytosis. Cancer. 1979; 44: 993-1002.
- Hadchouel M, Prieur AM, Griscelli C. Acute hemorrhage, hepatic, and neurologic manifestations in juvenile rheumatoid arthritis: Possible relationship to drugs or infection. J Pediatr. 1985;106:561-566.
- Sawhney S, Won P, Murray KJ. Macrophage activation syndrome: A potentially fatal complication of rheumatic disorders. Arch Dis Child. 2001;85:421-426.
- Blessing J, Prada A, Siegel DM, et al. The diagnostic significance of soluble CD163 and soluble interleukin-2 receptor alpha-chain in macrophage activation syndrome and untreated new-onset systemic juvenile idiopathic arthritis. Arthritis Rheum. 2007;56:965-971.
- Behrens EM, Beukelman T, Paessler M, Cron RQ. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol. 2007;34:1133-1138.