
Why do chronic inflammatory diseases (CIDs) such as rheumatoid arthritis (RA) or systemic lupus erythematosus affect the whole body and produce myriad debilitating and disabling symptoms that make people sick? Is this clinical pattern of systemic involvement an unfortunate byproduct of sustained inflammation or, in an unexpected way, is it an adaptive program positively selected during evolution?
The systemic disease sequelae in CIDs are many and represent some of the major problems for which patients seek medical attention and require treatment. These sequelae include the following:
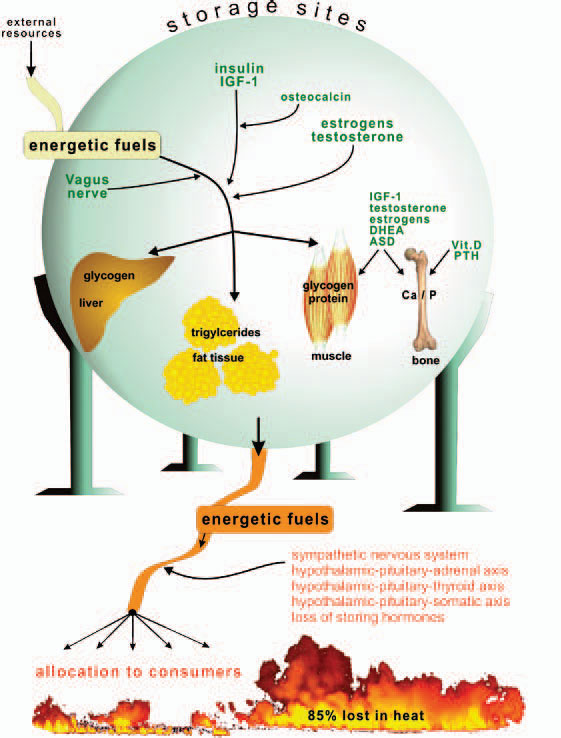
Storage of energy is maximal when resources from the food supply are available ad libitum and energy consumers such as brain, muscles, and others are minimally active. Note that the central nervous system, muscles, and the immune system are the major energy consumers in the body, with each utilizing approximately 2,000–2,500 kJ/day.
Storage of energetic fuels is mainly supported by anabolic pathways such as the parasympathetic nervous system with the vagus nerve (uptake in the gut and liver), insulin from the pancreas (uptake of glucose into liver, muscle, and fat tissue and storage of triglycerides), insulin-like growth factor–1 (for growth support), estrogens (support of fat tissue and growth), androgens (support of muscle tissue and growth), vitamin D and parathyroid hormone (support of bone and calcium and phosphate storage), and osteocalcin (support of insulin actions).1 Figure 1 (p. 25) summarizes neuroendocrine regulation of storage of energetic substrates.
Provision of energetic fuels is mainly mediated by catabolic pathways such as those regulated by the sympathetic nervous system (SNS; via β2-adrenoceptors, gluconeogenesis, and lipolysis); the hypothalamic-pituitary-adrenal (HPA) axis (cortisol, gluconeogenesis, and protein breakdown in muscles); and the hypothalamic-pituitary-thyroid axis (thyroid hormones support effects of the SNS) (see Figure 1, p. 25).
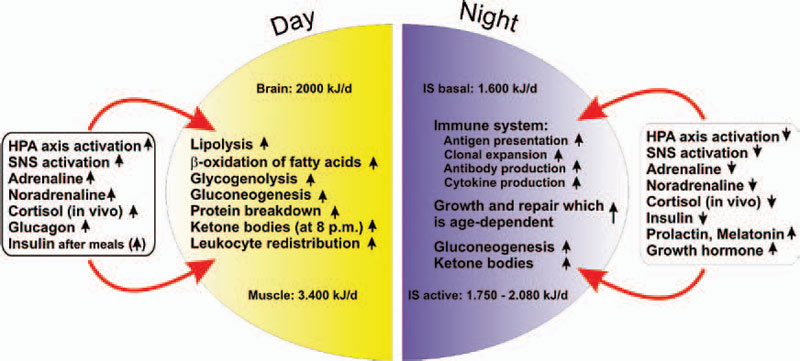
If all bodily organs consumed limiting resources simultaneously, a serious shortage would rapidly ensue, and insufficient energy would be available to sustain organ function. Regulation of energy allocation to stores or consumers is a delicate process. Indeed, the brain and muscles mainly need energetic fuels when active during the day, while immune system activity and growth processes are augmented during night. Neuroendocrine regulation of energy allocation follows a circadian rhythm (see Figure 2, p. 25).1 An important question thus arises: Have complex neuroendocrine immune interactions been positively selected during evolution in the context of CIDs, or do they have another basis?
Evolutionary Medicine
In the pathogenesis of symptomatic CIDs, genes that might play a specific role (favorable or unfavorable) may not have escaped conservation evolutionarily because unrestricted gene transmission was not possible for the following reasons:9
- High negative selection pressure. Thus, CIDs may lead to loss of reproducibility because affected individuals are at a disadvantage, excluded, or impaired in the competition for food, positions in the group, and sexual partners. In addition, long-term high-inflammatory activity may inhibit the hypothalamic-pituitary-gonadal axis, leading to impairment of fertility.10-14
- No selection pressure at all. At present, many CIDs only become manifest in patients at older ages. Due to the short life expectancy in the past, our ancestors may not have suffered from the CIDs that we know today.
- There was no time for natural selection. This situation would occur if various CIDs did not exist 100 to 200 years ago. Indeed, there is a suggestion that RA, for example, is a relatively new disease.
Nevertheless, genes can be transmitted before the outbreak of a CID and persist among descendants. On the other hand, genes whose products operate in the symptomatic phase of CIDs will not be positively selected due to negative influence of the disease (negative selection pressure). Transmitted genes may confer an increased risk for a CID, but, most likely, these genes were positively selected for fitness in reproduction and survival at younger ages independent of the CID (antagonistic pleiotropy).15 For example, HLA DR4 (DRB1*04), known to be a risk factor in RA, shows a high negative association with the risk of dengue hemorrhagic fever.16 It is possible, therefore, that HLA-DR4 (DRB1*04) was positively selected to fight off dengue hemorrhagic fever (or some other infection), although later in life, this particular surface molecule supports the development of a CID by increasing antigen presentation and T cell activation.9,17
It make sense that specific genes have not been selected or conserved in evolution because of their putative role in CIDs, either positive or negative. Rather, genes, signaling pathways, and networks operative in symptomatic CIDs were likely borrowed from serious, albeit non-life-threatening, inflammatory episodes, such as transient events. Recovery is possible from these episodes, but they would not have blocked the gene transmission to the progeny. According to these ideas, genes and the response programs they regulate are conserved for host defense against infection, control of inner and outer body surfaces, foreign body reactions, wound healing and repair, immunosurveillance in tissue, implantation of the stem cells into injured tissue, immune phenomena facilitating semiallogenic pregnancy, replacement of cells and tissue (physiological regeneration and degeneration), and similar ones. The same is true for neuroendocrine regulation of energy storage and energy allocation to consumers.
Energy Regulation and Evolutionary Medicine
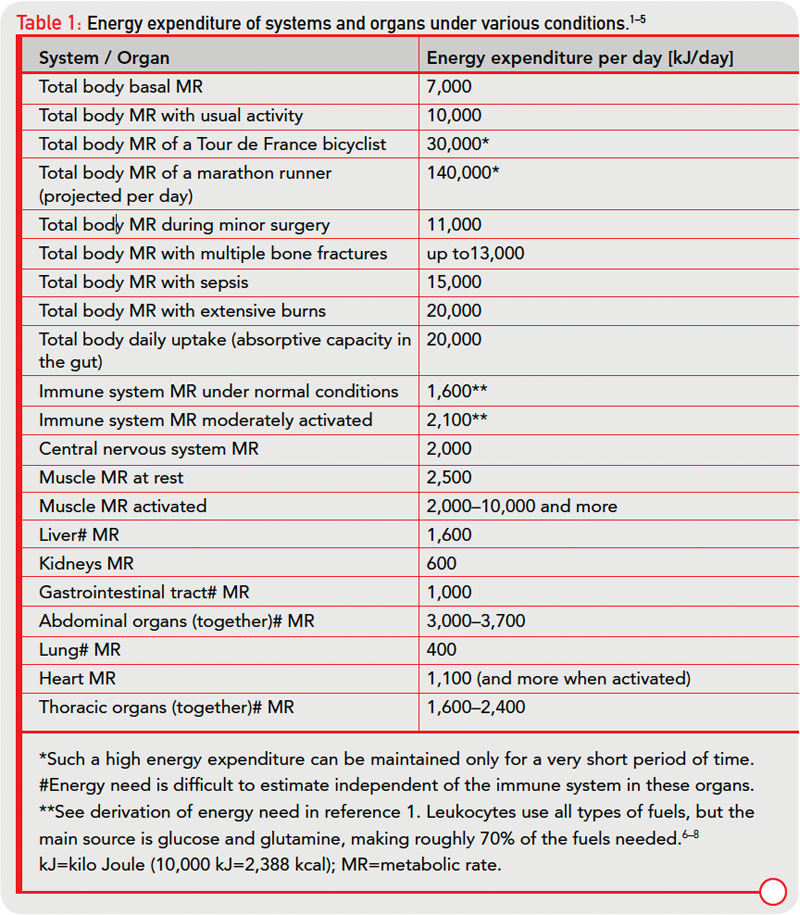
In an acute inflammatory infectious disease, the immune system has a pressing need to boost its energy supply (see Table 1, at right). To meet this demand, neuroendocrine systems are switched on to provide energy-rich substrates to the activated immune system (the red hormone axes in Figure 1, p. 25). Most likely, these responses have been evolutionarily conserved for acute inflammatory episodes.
Any change in energy availability can happen only when the inflammatory process switches on a systemic neuroendocrine response. If an inflammatory process is restricted to a local tissue compartment, no systemic response is induced, and local energy resources are used to nourish activated immune cells.1 Fortunately, in this situation, the disease is confined to a local process. In contrast, if the inflammatory process is strong, a systemic response is required to induce systemic energy allocation to the activated immune system.1 Several pathways exist for stimulation of neuroendocrine centers in the brain by the activated immune system.18
As it persists, the inflammatory disease process enters a systemic stage to appeal for remote energy resources (glucose from the liver, amino acids from distant muscles, free fatty acids from distant fat, with the help of calcium and phosphorus from remote bone) (see Figure 1, p. 25). The presented theory states that this appeal or “energy appeal reaction” has been evolutionarily conserved as an adaptive program for transient inflammatory episodes of a systemic nature. In the context of a CID, however, the continuous energy appeal reaction leads to typical systemic disease sequelae.
Depressive Symptoms and Fatigue: Sickness Behavior
Fatigue and related depression appear to be part of an adaptive program that operates during short-lived events such as infection to limit activities of the brain and muscles. This program provides energy-rich fuels for the activated immune system and confines the affected person to a safe place. Mechanisms of sickness behavior have been described for over three decades.19
Anorexia and Malnutrition
Anorexia is a consequence of sickness behavior and is necessary to conserve energy because foraging behavior is energy consuming. Anorexia is a continuous problem in CIDs (see references in Reference 1). Unfortunately, long-term anorexia leads to malnutrition as demonstrated in CIDs.1 Importantly, restricted intake of food can lead to deficiencies of vitamin D and vitamin K, as well as deficiencies of metal ions such as zinc, iron, copper, and magnesium, among others.
Muscle Wasting, Cachexia, and Cachectic Obesity
Cachexia is frequent in the context of CIDs.20,21 The basis of cachexia is at least twofold: 1) proinflammatory cytokines impair muscle growth (increased levels of myostatin); and 2) chronic loss of androgens promotes muscle wasting. In this setting, muscle proteins are degraded and the resulting amino acids are used in gluconeogenesis in the liver. During transient episodes of inflammation under conditions of anorexia, this change can help provide glucose and amino acids to an activated immune system. In addition, cachexia reduces time and energy spent by muscular work.
During starvation (think of the transient inflammatory episode that was the basis of evolutionary conservation), metabolic support for survival involves first glycogen stores in the liver (for half a day), then protein stores in the muscles (one to three days), and finally fat stores in adipose tissue (from day three on). By utilizing all reserves, a patient of normal weight can live up to 2.5 months. In the starvation program, liver glycogen and muscle protein are first used. If the full starvation program is not started after three days, fat reserves will be spared, but proteins from muscles will be mainly utilized (recall that free fatty acids can not be used in gluconeogenesis). Since cachectic obesity with muscle loss and maintenance of adipose tissue is a hallmark of many CIDs, such an incomplete starvation program is most probably active in CIDs.21
Insulin Resistance with Hyperinsulinemia
Resistance to insulin affects the liver (reduced glucose uptake), muscle (reduced glucose uptake), and fat tissue (reduced glucose uptake and reduced triglyceride storage). However, the activated immune system does not become insulin resistant. In contrast, insulin upregulates glucose transporters on immune cells that support glucose uptake. Insulin resistance is an adaptive program for short-lived inflammatory episodes to redirect glucose and free fatty acids to activated immune cells and to stimulate immune responses.1 In other words, the energy highways to liver, muscle, and fat tissue are detoured to the activated immune system.
Dyslipidemia
Although the pattern of dyslipidemia may vary between different CIDs, a common phenomenon in all CIDs is a low level of high-density lipoprotein (HDL) cholesterol and/or apolipoprotein A-I.1 HDL is instrumental in removing cholesterol from tissue (reverse cholesterol transport). Loss of typical HDL cholesterol and appearance of a proinflammatory form of HDL with decreased apolipoprotein A-I and increased serum amyloid A and ceruloplasmin is augmented in CIDs.22 In the context of acute inflammatory episodes, the CID-related transition of normal HDL to proinflammatory HDL becomes understandable as part of an adaptive program.23 For energy regulation, this response is appropriate because it inverts reverse cholesterol transport and, thus, increases provision of energy-rich fuels to immune cells (think of the fat-laden macrophage).
Increase of Adipose Tissue in the Proximity of Inflammatory Lesions
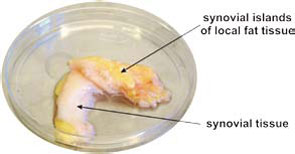
Many CIDs demonstrate fat deposits near inflammatory lesions such as creeping fat in Crohn’s disease or fat tissue adjacent to synovial tissue in RA (see Figure 3, p. 26). Because fibroblasts can easily transform into adipocytes, the appearance of fat tissue in the proximity of inflammatory lesions may be an adaptive program.24 Although several factors could lead to this type of transformation, locally increased estrogen levels could be an important mediator of this process.25 These fat deposits can nourish nearby inflammatory processes because immune cells directly use free fatty acids. This possibility is supported by an increased density of sympathetic nerve fibers in the juxtasynovial fat tissue of RA patients due to β2‑adrenergically induced lipolysis.26
Alterations of Steroid Hormone Axes
The well-known mild increase of the HPA axis activity in CIDs (albeit inadequately low to suppress chronic inflammation) with elevated cortisol levels is an adaptive program to support gluconeogenesis in the liver. In addition, this program redirects energy-rich fuels to the activated immune system by catabolic influences on muscle (protein degradation, amino acids for gluconeogenesis).
Similarly, the loss of adrenal and gonadal hormones belongs to an adaptive program because reproduction is switched off at the expense of immune defense mechanisms.27,28 The whole reproductive program, including courtship behavior, is too energy consuming. In addition, the loss of androgens is an important factor to increase muscle wasting and, thus, re-allocation of muscle proteins to gluconeogenesis.
Elevated Sympathetic Tone
Increased sympathetic activity has been described in CIDs, although the meaning is unclear.1,29 Increased sympathetic activity leads to gluconeogenesis and, more importantly, to β2‑adrenergically mediated lipolysis. In this situation, both pathways may be switched on to support an activated immune system, but its continuous operation is unfavorable in CIDs. The loss of sympathetic nerve fibers described in inflammatory lesions is another hallmark of tissue inflammation. Importantly, sympathetic nerve fibers appear in high density in the fat tissue surrounding inflammatory lesions and activated lymph nodes in arthritis.26 This development supports lipolysis in neighboring fat tissue.
Decreased Parasympathetic Tone
Consistent with an increased activity of the SNS, decreased activity of the parasympathetic nervous system can occur during CIDs.30 The SNS is switched on at the expense of the parasympathetic system, which is linked to reduced gastrointestinal activity, decreased nutrient uptake, and diminished glucose uptake into the liver. As in the case of other systemic events described, the program may have been evolutionarily conserved for transient inflammatory episodes but becomes unfavorable in CIDs because the antiinflammatory cholinergic pathway is not active.31
Inflammation-Related Anemia
The following causes of anemia have been described in disease pathology: 1) allocation of iron to macrophages and other cells of the reticuloendothelial system stimulated by hepcidin; 2) reduced intestinal iron resorption initiated by hepcidin; 3) disturbed erythropoiesis due to proinflammatory cytokines and reduced half-life of erythrocytes (phagocytosis by macrophages); and 4) reduced influence of erythropoietin on erythropoiesis (little production or resistance). Circulating cytokines stimulate these pathophysiological elements.
For overall redirection of energy-rich substrates to the activated immune system, this program has the following significance: 1) Anemia is accompanied by reduced energy expenditure for erythropoiesis (events during sickle-cell anemia depict how much energy is needed for red blood cell production32); 2) lower oxygen-transport capacity and lower oxygen tension leads to decreased physical activity; 3) reduced iron levels are followed by decreased myoglobin levels in skeletal muscle (lower muscular work); and 4) absorbed iron is used in activated macrophages (necessary for peroxide- and nitric oxide–generating enzymes [usually for killing microbes], and used for cytochromes in the respiratory chain complex to handle necessary protons for adenosine triphosphate production). From this point of view, mild CID-related anemia is an adaptive program.
Osteopenia
Calcium as well as phosphorus are essential for energy consumption.33 To meet metabolic demands during CIDs, both elements need to be provided from bone (internal store) because the fatigue, anorexia, and malnutrition that accompany inflammatory disease lead to reduced calcium and phosphorus uptake in the gut (think of the starvation program).
Several important factors contribute to osteopenia in CIDs: 1) vitamin D and K deficiency (see the above section on anorexia); 2) magnesium deficiency (see the above section on anorexia); 3) upregulation of proinflammatory cytokines supporting osteoclast activity via RANKL; 4) increased SNS activity34; 5) increased cortisol levels35; and 6) decreased gonadal hormones. Together, these changes can promote osteoclastic bone resorption while osteoblastic bone generation is inhibited.
Recently, the role of osteocalcin, an osteoblast hormone, has been revisited.36 Studies have shown that this hormone increases insulin sensitivity, indicating that bone-sparing factors such as osteocalcin can support glucose uptake and fuel storage. Although these metabolic changes may occur in CIDs and influence pathogenesis, they likely have been evolutionarily conserved for transient inflammatory episodes.
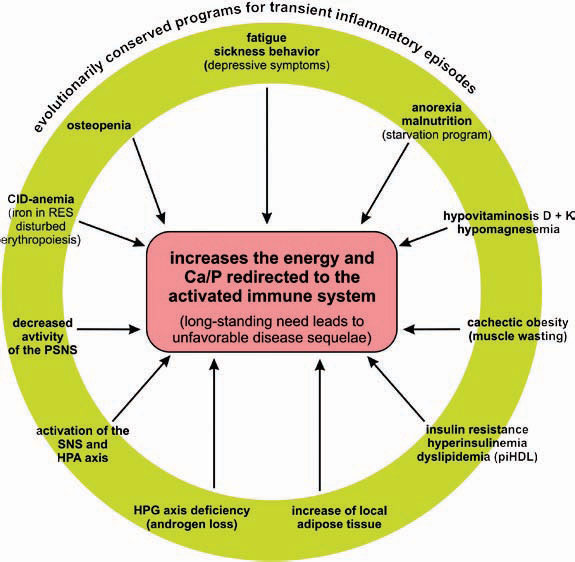
Conclusion
In the past, several notable features of CIDs were considered independent phenomena in the absence of a unifying model to explain both their prominence and coexistence. As a common denominator, the redirection of energy-rich fuels from energy stores to the activated immune system can explain systemic disease sequelae (see Figure 4, above). This theory also can explain the observed disturbances of the neuroendocrine immune regulation in CIDs. In this conceptualization, the induction of systemic responses results from an appeal for energy-rich substrates, called the energy appeal reaction. The theory states that the energy appeal reaction has been positively selected for transient inflammatory episodes, but prolonged use of adaptive programs can become pathogenic. Thus, one can add the “systemic response” to the list of classical etiologic factors of CIDs (see Table 2, p. 26). Hopefully, recognition of the importance of energy fluxes during both acute and chronic inflammation can provide a new perspective of disease pathogenesis and lead to better approaches for diagnosis and treatment.
Acknowledgments
The theory demonstrated in this paper was developed by the author during the last 10 years in individual steps that have been published previously.1,9,17,37
Dr. Straub is a professor in the Laboratory of Experimental Rheumatology and Neuroendocrine Immunology in the Department of Internal Medicine I, University Hospital in Regensburg, Germany.
References
- Straub RH, Cutolo M, Buttgereit F, Pongratz G. Energy regulation and neuroendocrine-immune control in chronic inflammatory diseases. J Intern Med. 2010;267:543-560.
- Blaxter K. Energy metabolism in animals and man. Cambridge: Cambridge University Press; 1989.
- Rassow J, Hauser K, Netzker R, Deutzmann R. Biochemistry. Stuttgart, Germany: Georg Thieme; 2008.
- Iemitsu M, Itoh M, Fujimoto T, Tashiro M, et al. Whole-body energy mapping under physical exercise using positron emission tomography. Med Sci Sports Exerc. 2000;32:2067-2070.
- Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev. 1997;77:731-758.
- Calder PC. Fuel utilization by cells of the immune system. Proc Nutr Soc. 1995;54:65-82.
- Maciver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL, Rathmell JC. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J Leukoc Biol. 2008;84:949-957.
- Frauwirth KA, Thompson CB. Regulation of T lymphocyte metabolism. J Immunol. 2004;172:4661-4665.
- Straub RH, Besedovsky HO. Integrated evolutionary, immunological, and neuroendocrine framework for the pathogenesis of chronic disabling inflammatory diseases. FASEB J. 2003;17:2176-2183.
- Ostensen M, Almberg K, Koksvik HS. Sex, reproduction, and gynecological disease in young adults with a history of juvenile chronic arthritis. J Rheumatol. 2000;27:1783-1787.
- Silva CA, Deen ME, Febronio MV, et al. Hormone profile in juvenile systemic lupus erythematosus with previous or current amenorrhea. Rheumatol Int. 2010 Mar 21 [Epub ahead of print].
- Villiger PM, Caliezi G, Cottin V, Forger F, Senn A, Ostensen M. Effects of TNF antagonists on sperm characteristics in patients with spondyloarthritis. Ann Rheum Dis. 2010;69:1842-1844.
- Soares PM, Borba EF, Bonfa E, Hallak J, Correa AL, Silva CA. Gonad evaluation in male systemic lupus erythematosus. Arthritis Rheum. 2007;56:2352-2361.
- Suehiro RM, Borba EF, Bonfa E, et al. Testicular Sertoli cell function in male systemic lupus erythematosus. Rheumatology (Oxford). 2008;47:1692-1697.
- Williams GC. Pleiotropy, natural selection, and the evolution of senescence. Evolution. 1957;11:398-411.
- LaFleur C, Granados J, Vargas-Alarcon G, et al. HLA-DR antigen frequencies in Mexican patients with dengue virus infection: HLA-DR4 as a possible genetic resistance factor for dengue hemorrhagic fever. Hum Immunol. 2002;63:1039-1044.
- Straub RH, Del Rey A, Besedovsky HO. Emerging concepts for the pathogenesis of chronic disabling inflammatory diseases: Neuroendocrine-immune interactions and evolutionary biology. In: R Ader, ed. Psychoneuroimmunology. San Diego, Calif.: Elsevier Academic Press; 2007:217-232.
- Besedovsky HO, Del Rey A. Immune-neuro-endocrine interactions. Endocr Rev. 1996;17:64-102.
- Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46-56.
- Roubenoff R, Roubenoff RA, Cannon JG, et al. Rheumatoid cachexia: Cytokine-driven hypermetabolism accompanying reduced body cell mass in chronic inflammation. J Clin Invest. 1994;93:2379-2386.
- Walsmith J, Roubenoff R. Cachexia in rheumatoid arthritis. Int J Cardiol. 2002;85:89-99.
- Hahn BH, Grossman J, Ansell BJ, Skaggs BJ, McMahon M. Altered lipoprotein metabolism in chronic inflammatory states: Proinflammatory high-density lipoprotein and accelerated atherosclerosis in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Res Ther. 2008;10:213.
- Khovidhunkit W, Kim MS, Memon RA, et al. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res. 2004;45:1169-1196.
- Schäffler A, Müller-Ladner U, Schölmerich J, Büchler C. Role of adipose tissue as an inflammatory organ in human diseases. Endocr Rev. 2006;27:449-467.
- Cutolo M, Straub RH, Bijlsma JW. Neuroendocrine-immune interactions in synovitis. Nat Clin Pract Rheumatol. 2007;3:627-634.
- Straub RH, Lowin T, Klatt S, Wolff C, Rauch L. Increased density of sympathetic nerve fibers in metabolically activated fat tissue surrounding human synovium and mouse lymph nodes in arthritis. Arthritis Rheum. 2011 Jun 23 [Epub ahead of print].
- Tsigos C, Papanicolaou DA, Kyrou I, Raptis SA, Chrousos GP. Dose-dependent effects of recombinant human interleukin-6 on the pituitary-testicular axis. J Interferon Cytokine Res. 1999;19:1271-1276.
- Rivier C. Role of endotoxin and interleukin-1 in modulating ACTH, LH and sex steroid secretion. Adv Exp Med Biol. 1990;274:295-301.
- Dekkers JC, Geenen R, Godaert GL, Bijlsma JW, van Doornen LJ. Elevated sympathetic nervous system activity in patients with recently diagnosed rheumatoid arthritis with active disease. Clin Exp Rheumatol. 2004;22:63-70.
- Sloan RP, McCreath H, Tracey KJ, Sidney S, Liu K, Seeman T. RR interval variability is inversely related to inflammatory markers: The CARDIA study. Mol Med. 2007;13:178-184.
- Rosas-Ballina M, Tracey KJ. Cholinergic control of inflammation. J Intern Med. 2009;265:663-679.
- Hibbert JM, Creary MS, Gee BE, Buchanan ID, Quarshie A, Hsu LL. Erythropoiesis and myocardial energy requirements contribute to the hypermetabolism of childhood sickle cell anemia. J Pediatr Gastroenterol Nutr. 2006; 43:680-687.
- Feske S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol. 2007;7:690-702.
- Takeda S, Karsenty G. Molecular bases of the sympathetic regulation of bone mass. Bone. 2008;42:837-840.
- Duncan H. Osteoporosis in rheumatoid arthritis and corticosteroid induced osteoporosis. Orthop Clin North Am. 1972;3:571-583.
- Lee NK, Karsenty G. Reciprocal regulation of bone and energy metabolism. Trends Endocrinol Metab. 2008; 19:161-166.
- Straub RH. Concepts of evolutionary medicine and energy regulation contribute to the etiology of systemic chronic inflammatory diseases. Brain Behav Immun. 2011;25:1-5.
