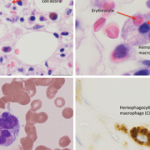
Because expansion of tissue macrophages, or histiocytes, exhibiting hemophagocytic activity in MAS is often triggered by infections or modifications in the drug therapy, the term reactive hemophagocytic lymphohistiocytosis has been preferred by some authors to classify this condition.6 This term suggests that MAS may belong to a group of histiocytic disorders collectively known as hemophagocytic lymphohistiocytosis (HLH). HLH is a more general term that describes a spectrum of diseases characterized by accumulations of histologically benign well-differentiated mononuclear cells with a macrophage phenotype.7-9 Because such macrophages represent a subset of histiocytes that are distinct from Langerhans cells, this entity should be distinguished from Langerhans cell histiocytosis as well as other dendritic cell disorders. In the current classification of histiocytic disorders, HLH is further subdivided into primary or familial HLH and secondary or reactive HLH. Clinically, however, these conditions may be difficult to distinguish from each other. Familial hemophagocytic lymphohistiocytosis (FHLH) is a constellation of rare autosomal recessive immune disorders. The clinical symptoms of FHLH usually become evident within the first two months of life. The course of disease is very severe, and most patients eventually require hematopoietic stem cell transplantation. Secondary HLH tends to occur in older children and more often is associated with an identifiable infectious episode, most notably Epstein-Barr virus (EBV) or cytomegalovirus (CMV) infection. The group of secondary hemophagocytic disorders also includes malignancy associated HLH. Although there are many clinical similarities between MAS and both primary and secondary HLH, the exact relationship between these conditions is not understood.
Pathophysiology
The pathological mechanisms of MAS are not fully understood. In clinically similar primary HLH, the uncontrolled proliferation of T cells and macrophages has been linked to decreased natural killer (NK) cell and cytotoxic T cell function, often due to mutations in the gene encoding perforin.8,10 Perforin is a protein that cytolytic cells utilize to induce apoptosis of target cells, such as tumor cells or cells infected by viruses. In a sizable proportion of patients with primary HLH, the disease is caused by mutations in another gene, MUNC13-4.11 The protein encoded by the MUNC13-4 gene is involved in the release of perforin into the immune synapse with a target cell. Although the cytolytic cells of patients with MUNC13-4 mutations produce sufficient amounts of perforin, their ability to kill target cells is greatly diminished. Defects in the granule-dependent cytotoxic functions of lymphocytes have also been implicated in several other genetic diseases associated with the hemophagocytic syndrome.
Recent observations suggest that as in HLH, MAS patients have profoundly depressed NK cell function, often associated with abnormal perforin expression, and these abnormalities are associated with specific MUNC13-4 and perforin gene polymorphisms.12,13 Although the exact pathways that could link cytolytic abnormalities in these patients with excessive expansion of macrophages are not clear, the presence of defects in the granule-dependent cytotoxic activity of lymphocytes in several diseases associated with hemophagocytic syndromes highlights the importance of this function in restoring the immune system to a state of equilibrium during some inflammatory responses.
Two possible scenarios explaining how cytolytic dysfunction may lead to excessive activation of macrophages have been suggested in the literature. One is related to the fact that HLH/MAS patients appear to have a diminished ability to control some infections.14 More specifically, in this scenario, NK cells and cytotoxic T lymphocytes (CTLs) fail to kill infected cells and thus remove the source of antigenic stimulation. Such persistent antigen stimulation leads, in turn, to persistent antigen-driven activation and proliferation of T cells associated with persistent production of cytokines that stimulate macrophages. A second scenario is based on the observations that, in some circumstances, cytotoxic cells may be directly involved in induction of apoptosis of activated macrophages and T cells during the contraction stage of the immune response.15 In both scenarios, cytotoxic dysfunction leads to persistent expansion of T cells and macrophages and escalating production of proinflammatory cytokines. Indeed, the clinical findings during the acute phase of MAS can largely be explained as a consequence of the prolonged production of cytokines and chemokines.
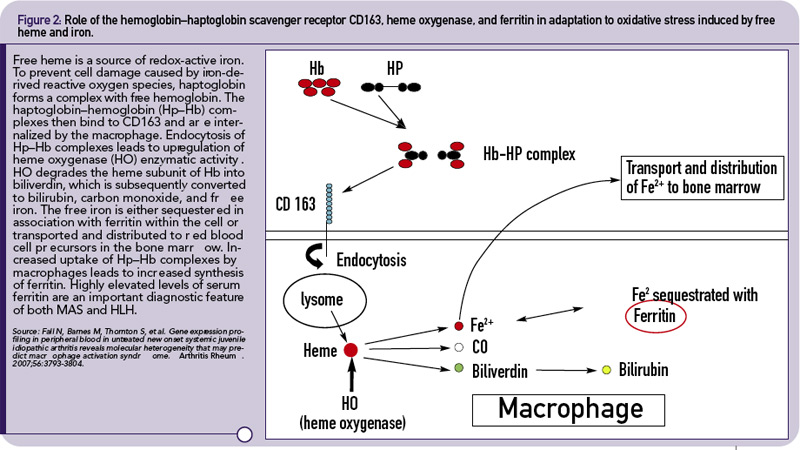
The hemophagocytic macrophages in MAS express CD163.16,17 This feature provides clues to the understanding of the origin of extreme hyperferritinemia in MAS. The only proven function of CD163 is related to its ability to bind hemoglobin– haptoglobin complexes and initiate pathways important for the adaptation to oxidative stress induced by free heme and iron (see Figure 2, p 29).18 In these pathways, to prevent cell damage caused by iron-derived reactive oxygen species, free hemoglobin, the main source of redox-active iron, forms a complex with haptoglobin. The haptoglobin–hemoglobin (Hp–Hb) complexes then bind to CD163 and are internalized by the macrophage. Endocytosis of Hp–Hb complexes leads to upregulation of heme oxygenase (HO) enzymatic activity. HO degrades the heme subunit of Hb into biliverdin, which is subsequently converted to bilirubin, carbon monoxide and free iron. The free iron is either sequestered in association with ferritin within the cell or transported and distributed to red blood cell precursors in the bone marrow. Increased uptake of Hp–Hb complexes by macrophages leads to increased synthesis of ferritin. In MAS, increased release of free Hb associated with erythrophagocytosis would require increased production of ferritin to sequestrate an excessive amount of free iron. Indeed, a strikingly high level of serum ferritin (often above 10,000 ng/dL) is an important diagnostic feature of both MAS and HLH.
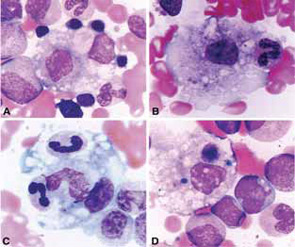
Clinical Features
The clinical findings in overt MAS are often dramatic and evolve rapidly. Typically, patients with a chronic condition become acutely ill with persistent fever, mental status changes, lymphadenopathy, hepatosplenomegaly, and liver dysfunction. A hemorrhagic syndrome resembling disseminated intravascular coagulopathy is another striking feature of MAS. Hemorrhagic skin rashes from mild petechiae to extensive ecchymotic lesions, epistaxis, hematemesis secondary to upper gastrointestinal bleeding, and rectal bleeding, are commonly seen in these patients. These clinical symptoms are associated with a precipitous fall in at least two of three blood cell lines (leukocytes, erythrocytes, and platelets). The fall in platelet count is usually an early finding. Because bone marrow aspiration typically reveals significant hypercellularity and normal megakaryocytes, such cytopenias do not seem to be secondary to inadequate production of cells. Increased destruction of the cells by phagocytosis and consumption at the inflammatory sites are more likely explanations.
A precipitous fall in erythrocyte sedimentation rate (ESR) despite persistently high C-reactive protein is another characteristic laboratory feature, which probably reflects the degree of hypofibrinogenemia secondary to fibrinogen consumption and liver dysfunction. In fact, liver involvement is common in MAS. Significant hepatomegaly is frequently present. Some patients develop mild jaundice. Liver function tests often reveal high serum transaminases activity and mildly elevated levels of serum bilirubin. Moderate hypoalbuminemia has been reported as well. Serum ammonia levels are typically normal or only mildly elevated.
Encephalopathy is another frequently reported clinical feature of MAS. Mental status changes, seizures, and coma are the most common manifestations of central nervous system disease. Cerebrospinal fluid pleiocytosis with mildly elevated protein has been noted in some studies. Significant deterioration in renal function has been noted in several series and was associated with particularly high mortality in one report.3 Pulmonary infiltrates have been mentioned in several reports, and hemophagocytic macrophages can be found in bronchoalveolar lavage.
Additional laboratory findings in MAS include highly elevated serum levels of triglycerides and ferritin. The elevation of ferritin is particularly marked (above 10,000 ng/mL) in most patients and appears to parallel the degree of macrophages activation.
Diagnosis
There are no validated diagnostic criteria for MAS, and early diagnosis is often difficult. Thus, in patients with persistently active underlying rheumatologic disease, a fall in the ESR and platelet count, particularly in a combination with increase in serum D-dimer and ferritin levels, should raise a suspicion of impending MAS. The diagnosis of MAS is usually confirmed by the demonstration of hemophagocytosis in the bone marrow. However, the demonstration of hemophagocytosis in the bone marrow may be difficult due to sampling errors, particularly at the early stages of the syndrome. In addition, it is now increasingly recognized that hemophagocytic macrophages may infiltrate tissues other than bone marrow (e.g., liver, lymph nodes, skin, and lungs). In contrast to MAS, the diagnosis of HLH is usually established based on the validated diagnostic criteria developed by the International Histiocyte Society.9 The criteria include either:
- A molecular diagnosis based on specific mutations found in either PRF1 or MUNC13-4 genes; or
- A clinical diagnosis based on the following clinical criteria:
- Persistent fever;
- Splenomegaly;
- Cytopenias involving at least two cell lines;
- Hypertrigliceremia and/or hypofibrinogenemia;
- Hyperferritinemia; and
- Hemophagocytosis in the bone marrow.
More recently, two additional laboratory criteria have been added:
- Low/absent NK cell cytolytic activity; and
- High sIL2Rα levels.
The definite clinical diagnosis of HLH requires the presence of at least five of the eight criteria.
Unfortunately, the application of the HLH diagnostic criteria to systemic JIA patients with suspected MAS is problematic. Some of the HLH markers such as lymphadenopathy, splenomegaly, and hyperferritinemia are common features of active SJIA itself and, therefore, do not distinguish MAS from a conventional SJIA flare. Other HLH criteria, such as cytopenias and hypofibrinogenemia, become evident only at the late stages. This is related to the fact that systemic JIA patients often have increased white blood cell and platelet counts as well as increased serum levels of fibrinogen as a part of the inflammatory response seen in this disease. Therefore, when these patients develop MAS, they may only reach the degree of cytopenias and hypofibrinogenemia seen in HLH at the late stages of the syndrome when their management becomes challenging. The application of criteria is even more problematic for the diagnosis of MAS in patients with SLE in whom autoimmune cytopenias are common and difficult to distinguish from those caused by MAS. In these patients, the presence of extreme hyperferritinemia and lactate dehydrogenase elevation should raise suspicion for MAS. Attempts to modify the HLH criteria to increase their sensitivity and specificity for the diagnosis of MAS in rheumatic conditions have been initiated.19
Differential Diagnosis
In addition to distinguishing MAS from a flare of an underlying rheumatologic disease, one must consider in differential diagnosis other clinical entities associated with hepatic dysfunction, coagulopathy, cytopenias, or encephalopathy. In some MAS patients, the combination of hepatic dysfunction with encephalopathy may be reminiscent of Reye syndrome. The diagnosis of Reye syndrome, however, is based of the presence of a viral prodrome, unexplained vomiting, behavioral changes, and a distinctive chemical profile characterized by rapid coordinated increase in serum aminotransferase levels, blood ammonia, and prothrombin time with relatively minimal changes in serum bilirubin. The DIC-like coagulopathy seen in MAS is not a feature of Reye syndrome. Conversely, a sharp increase in blood ammonia levels, an important feature of Reye syndrome, is usually very mild in MAS. The hemorrhagic syndrome seen in MAS may resemble thrombotic thrombocytopenic purpura. However, microangiopathic anemia with emergence of fragmented red blood cells in peripheral circulation, a central feature of thrombotic thrombocytopenic purpura, is usually not seen in MAS. It is also important to differentiate MAS from malignancy-associated HLH and malignant histiocytic disorders. Some other important differential diagnoses include sepsis and drug reactions.
Treatment
MAS is a life-threatening condition associated with high mortality rates. Therefore, early recognition and of this syndrome and immediate therapeutic intervention to produce a rapid response are critical. Most clinicians start with intraveneous methylprednisolone pulse therapy (30 mg/kg for three consecutive days) followed by 2–3 mg/kg/day in four divided doses. If a response to steroids is not evident within 24–48 hours, parenteral administration of cyclosporine A (CyA; 2–7 mg/kg/day) should be initiated.1,4,20 Patients in whom MAS remains active despite the use of corticosteroids and CyA present a serious challenge. In these patients, one might consider using the HLH-2004 treatment protocol developed by the International Histiocyte Society (see Table 1, p. 23). In addition to steroids and CyA, this protocol also includes etoposide (or VP16), a podophyllatoxin derivative that inhibits DNA synthesis by forming a complex with topoisomerease II and DNA. However, potential toxicity of the drug including severe myelosuppression is a major concern, particularly in patients with hepatic impairment. Etoposide is metabolized by the liver and then both the unchanged drug and its metabolites are excreted through the kidneys. Because patients who may require the use of etoposide are very likely to have hepatic and renal involvement, caution should be exercised to properly adjust the dosage and thus limit the extent of potential side effects such as severe bone marrow suppression,which may be detrimental. Reports describing deaths caused by severe bone marrow suppression and overwhelming infection have been published.
Recently it has been suggested that in patients unresponsive to the combination of steroids and CyA, particularly in those with renal and hepatic impairment, antithymocyte globulin (ATG) might be a safer alternative to etoposide.21 ATG depletes both CD4+ and CD8+ T cells through complement-dependent cell lysis. Mild depletion of monocytes is noted in some patients as well. Although in the reported cases this treatment was tolerated well, one must remember that infusion reactions are frequently reported with the use of ATG and adequate laboratory and supportive medical resources must be readily available if this treatment is used. The utility of biologic drugs in MAS treatment remains unclear. Although, tumor necrosis factor– (TNF-) inhibiting agents have been reported to be effective in occasional MAS patients, other reports describe patients in whom MAS developed while they were on TNF-inhibiting agents. Because, at least in systemic JIA, MAS episodes are often triggered by the disease flare, biologics that neutralize interleukin-1, a cytokine that plays a pivotal role in SJIA pathogenesis, have been tried by some authors. However, as with TNF-inhibiting agents, the results, however, have been conflicting. Based on some success of intravenous immunoglobin treatment in virus-associated reactive HLH, this treatment might be considered in MAS triggered by viral infection. If MAS, however, is driven by EBV infection, one might consider rituximab, a treatment that would eliminates B lymphocytes, the main type of cells harboring EBV virus. This approach has been successfully used in EBV-induced lymphoproliferative disease.22
Prognosis
Although the reported mortality rates of MAS reach 20%, due to increasing awareness it is now diagnosed relatively early and the outcome is improving. A substantial proportion of MAS patients experience recurrent episodes and these patients may require closer monitoring.
Dr. Grom is associate professor of pediatrics in the division of rheumatology at Cincinnati Children’s Hospital Medical Center.
References
- Hadchouel M, Prieur AM, Griscelli C. Acute hemorrhagic, hepatic, and neurologic manifestations in juvenile rheumatoid arthritis: Possible relationship to drugs or infection. J Pediatr. 1985;106:561-566.
- Grom AA. NK dysfunction: A common pathway in systemic onset juvenile rheumatoid arthritis, macrophage activation syndrome, and hemophagocytic lymphohistiocytosis. Arthritis Rheum. 2004; 50:689-698.
- Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: A potentially fatal complication of rheumatic disorders. Arch Dis Child. 2001; 85:421-426.
- Stephan JL, Kone-Paut I, Galambrun C, et al. Reactive haemophagocytic syndrome in children with inflammatory disorders: A retrospective study of 24 patients. Rheumatology (Oxford). 2001;40:1285-1292.
- Behrens EM, Beukelman T, Paessler M, Cron RQ. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol. 2007; 34:1133-1138.
- Athreya BH. Is macrophage activation syndrome is a new entity? Clin Exp Rheumatol. 2002; 20:121-123.
- Favara BE, Feller AC, Pauli M, et al. Contemporary classification of histiocytic disorders. The WHO Committee On Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of the Histiocyte Society. Med Pediatr Oncol. 1997;29:157-166.
- Filipovich HA. Hemophagocytic lymphohistiocytosis. Immunol Allergy Clin N Am. 2002;22:281-300.
- Henter JI, Horne A, Arico M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemopagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124-131.
- Stepp SE, Dufourcq-Lagelouse R, Le Deist F, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286:1957-1959.
- Feldmann J, Callebaut I, Raposo G, et al. MUNC13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell. 2003; 115:461-473.
- Grom AA, Villanueva J, Lee S, Goldmuntz EA, Passo MH, Filipovich AH. Natural killer cell dysfunction in patients with systemic-onset juvenile rheumatoid arthritis and macrophage activation syndrome. J Peds. 2003. 142;292-296.
- Vastert SJ, van Wijk R, D’Urbano LE, et al. Mutations in the perforin gene can be linked to macrophage activation syndrome in patients with systemic onset juvenile idiopathic arthritis. Rheumatology (Oxford). 2010;49:441-449.
- Menasche G, Feldmann J, Fischer A, de Saint Basile G. Primary hemophagocytic syndromes point to a direct link between lymphocyte cytotoxicity and homeostasis. Immunol Rev. 2005;203:165-179.
- Kagi D, Odermatt B, Mak TW. Homeostatic regulation of CD8+ T cells by perforin. Eur J Immunol. 1999;29:3262-3272.
- Avcin T, Tse SML, Schneider R, Ngan B, Siverman ED. Macrophage activation syndrome as the presenting manifestation of rheumatic diseases in childhood. J Pediatr. 2006; 148:683-686.
- Schaer DJ, Schleiffenbaum B, Kurrer M, et al. Soluble hemoglobin-haptoglobin scavenger receptor CD163 as a lineage-specific marker in the reactive hemophagocytic syndrome. Eur J Haemotol. 2005;74:6-10.
- Kristiansen M, Graversen JH, Jacobsen C, et al. Identification of the hemoglobin scavenger receptor. Nature. 2001;409:198-201.
- Ravelli A, Magni-Manzoni S, Pistorio A, et al. Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Pediatr. 2005;598-604.
- Ravelli A, De Benedetti F, Viola S, Martini A. Macrophage activation syndrome in systemic juvenile rheumatoid arthritis successfully treated with cyclosporine. J Pediatr. 1996;128:275-278.
- Coca A, Bundy KW, Marston B, Huggins J, Looney RJ. Macrophage activation syndrome: Serological markers and treatment with anti-thymocyte globulin. Clin Immunol. 2009; 132:10-18.
- Balamuth NJ, Nichols KE, Paessler M, Teachey DT. Use of rituximab in conjunction with immunosuppressive chemotherapy as a novel therapy for EBV-associated hemophagocytic lymphohistiocytosis. J Ped Hem/Oncol. 2007;8:569-573.