
There is a striking association of sJIA with secondary hemophagocytic lymphohistiocytosis, also known as macrophage activation syndrome (MAS). Although approximately 10% of sJIA patients develop overt MAS, up to 30% of children have evidence of subclinical MAS, characterized by a number of biochemical and immunological markers of MAS, including elevated levels of soluble CD163.2 In fact, some markers of MAS such as elevations of serum ferritin and d-dimers are considered typical laboratory manifestations of active systemic disease. It is interesting to note that both active sJIA and MAS are characterized by natural killer (NK) cell dysfunction. Full-blown MAS complicating sJIA is associated with significant morbidity and potentially mortality.
The following case represents a typical patient with early sJIA. We will present the case and discuss the treatment choices and outcome.
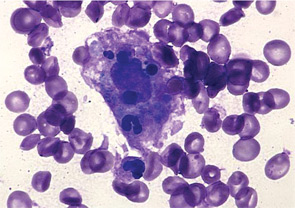
The Case
A 14-year-old boy was seen in the rheumatology clinic, having been unwell for approximately two months. He initially presented with diffuse arthralgias but subsequently developed fevers up to 102°F, usually once daily in the evening or early morning. He missed several days of school each week and estimated that he had lost 4 to 5 kg in weight. On physical examination, he had a typical systemic rash, diffuse lymphadenopathy, and oligoarthritis involving both wrists and one elbow. He did not have evidence of serositis or hepatosplenomegaly. After investigations excluded infections, malignancies, and other rheumatic diseases, a diagnosis of sJIA was made and he was treated with naproxen 500 mg bid. There was no improvement in his symptoms or laboratory markers when he was seen for a follow-up visit (see Table 2). Once the patient failed this initial nonsteroidal antiinflammatory drug (NSAID) trial, a decision had to be made about the next step in treatment.

