
Two new documents from the ACR were released this October, representing the latest data and consensus about systemic juvenile idiopathic arthritis (sJIA) and systemic sclerosis (SSc). The first document, which details treatment options for sJIA, is the first update of the ACR’s recommendations on pediatric rheumatic disease that were released two years ago. The second document, a joint effort of the ACR and the European League Against Rheumatism (EULAR), provides classification criteria for early disease in the subset of patients with SSc.
Recommendations for Three Phenotypes
The 2013 update gives treatment recommendations for three sJIA phenotypes, which were developed from an evaluation of 1,226 scenarios. The phenotypes are defined as: 1) active systemic features and varying degrees of synovitis; 2) no active systemic features and varying degrees of synovitis; and 3) systemic arthritis with features concerning for macrophage activation syndrome (MAS). Additionally, the document provides recommendations about repeat annual tuberculosis (TB) screening among children with JIA who, at baseline, had a negative TB evaluation.
“For those pediatric rheumatologists with a firm knowledge of the supporting literature on the treatment of sJIA, these recommendations may or may not affect their clinical practice,” says Pam Weiss, MD, MSCE, assistant professor and attending physician in the division of rheumatology at Children’s Hospital of Philadelphia. “But for pediatric rheumatologists less familiar with the literature, for adult rheumatologists caring for kids, and for general pediatricians without access to pediatric rheumatology colleagues, these guidelines should be a valuable resource for providing recommendations for the most common sJIA phenotypes that we see,” she says. Dr. Weiss and Sarah Ringold, MD, MS, assistant professor at the University of Washington School of Medicine and attending physician at Seattle Children’s Hospital, were principal investigators and coauthors of the new recommendations.
“We tried hard to find phenotypes that represent the most common presentations and cover the vast majority of patients that we see. But there clearly will be some sJIA patients that have complex phenotypes that are not covered with our groupings,” Dr. Weiss says.
Efficacy of IL-1, IL-6 Inhibitors
As with other categories of JIA, the goal of therapy for sJIA is “prompt control of active inflammation and symptoms and the prevention of a number of disease- and/or treatment-related morbidities such as growth disturbances, joint damage, and function limitations,” according to the document. Children with sJIA can have a particularly refractory course and persistent disease that leads to high risk of joint damage and severe growth impairment.
Because of the role that IL-1 and IL-6 play in the inflammatory process of sJIA, new drugs targeting those cytokines have been the focus of an increasing number of clinical trials. The new IL-1 and IL-6 inhibitors included in these recommendations are canakinumab, rilonacept, and tocilizumab.
When the first recommendations about JIA treatment were issued two years ago, IL-1 and IL-6 inhibitors had not been approved for sJIA. “Their approval and clinical data published since the last set of guidelines have had a profound impact on how we approach treatment of sJIA,” Dr. Weiss says.
Dr. Ringold agrees that recommendations about use of IL-6 and IL-1 inhibitors included in these new guidelines represent the most substantial change and should be particularly helpful for physicians who may not be aware of recent advances in treatment.
“A significant number of children are still cared for by adult rheumatologists who may be less familiar with the pediatric data. These recommendations will assist them,” Dr. Ringold says. Cost implications of these new therapies were not considered in development of the recommendations, per RAND/UCLA methodology.
Initial and Continuing Therapy
Treatment recommendations are listed for each of the phenotypes, and information is given about inappropriate use of certain therapies in certain situations. Among recommendations for patients with active systemic features and varying degrees of synovitis are ones about initiating anakinra, as well as systemic glucocorticoid (GC) monotherapy, either oral or intravenous, and nonsteroidal antiinflammatory drug (NSAID) monotherapy. Therapeutic options and indications are outlined for the treatment of patients within this category who have continued disease activity, including recommendations about use of abatacept, anakinra, a calcineurin inhibitor, and canakinumab, as well as when to initiate methotrexate, leflunomide, tocilizumab, or a tumor necrosis factor (TNF)–alpha inhibitor.
For patients without active systemic features and varying degrees of synovitis, intraarticular GC injection is recommended as an initial treatment option for certain patients, and methotrexate or leflunomide for others. Indications are given for use of NSAID monotherapy in patients without prior treatment. Therapeutic options for patients with continued disease activity include, under certain situations, abatacept, anakinra, canakinumab, tocilizumab, methotrexate or leflunomide, and a TNF-alpha inhibitor.
Initial therapeutic options for patients with sJIA with features concerning for MAS include anakinra, a calcineurin inhibitor, and systemic GC monotherapy. The authors note that development of treatment recommendations for this phenotype was challenging, given that there are no diagnostic or even classification criteria for MAS complicating sJIA and the etiology is probably heterogeneous. These recommendations will most likely be modified and expanded as more data about treatments become available.
Use of combination therapy with a biologic agent was not considered in the development of the recommendations and is not recommended because of concerns about safety and the lack of data. Any use of a biologic, therefore, should be part of a sequential therapy with prior biologic agents discontinued before starting a new one.
Dr. Ringold noted that the level of evidence for many of the recommendations is quite low. “I think some people may be surprised by the overall low level of evidence for some of the accepted approaches to therapy.” A higher level of evidence about efficacy of the new drugs, particularly the IL-1 and IL-6 inhibitors, is available, with data drawn from randomized trials, but often with small patient populations.
TB Screening
Public comment spurred inclusion of new recommendations about repeat TB screening for patients with sJIA. These new recommendations specify that annual screening is inappropriate for children at low risk of TB who had an initial negative TB test.
However, if the child’s TB risk changes to moderate or high, as determined by regional infectious disease guidelines, the screening should be repeated before beginning treatment with a biologic agent.
2013 SSc Classification Criteria
The 2013 Classification Criteria for Systemic Sclerosis, published in Arthritis & Rheumatism, represent an improvement on the 1980 ACR criteria, which classified diffuse SSc but did not include as many patients within the limited subset, resulting in at least 20% of patients with limited cutaneous disease not being classified correctly, according to Janet Pope, MD, MPH, a coauthor of the 2013 document and ACR principal investigator.2 Dr. Pope is professor of medicine in the division of rheumatology at the University of Western Ontario, Schulich School of Medicine.
The 1980 criteria “did not classify as many patients with early disease. There have also been advances in scleroderma: specific antibodies and nailfold capillary abnormalities that were not in the original criteria,” Dr. Pope says.
Developed by data collection and consensus among SSc experts from many countries in Europe and North America, the new classification system is based on clinical data and expert clinical judgment. The classification was tested repeatedly, including use of SSc cases and non-SSc controls, with the classification criteria then tested in a validation cohort and against preexisting criteria. The aims were to address a broader spectrum of SSc, including patients in the early stage of the disease; to include vascular, immunologic, and fibrotic manifestations of the disease; and to create criteria that can be used in research and clinical practice and would also be similar to diagnostic criteria if tested on early patients in the future, Dr. Pope says.
“Classification criteria are not diagnostic criteria, although they should be close,” Dr. Pope says. “The criteria help to classify more patients with SSc, where an expert would say the person has SSc, so that patients can potentially be classified earlier and then screened for important organ involvement.
“The real advance, however, is including more patients with SSc in clinical research who now meet the new criteria but did not previously meet the old criteria,” she says.
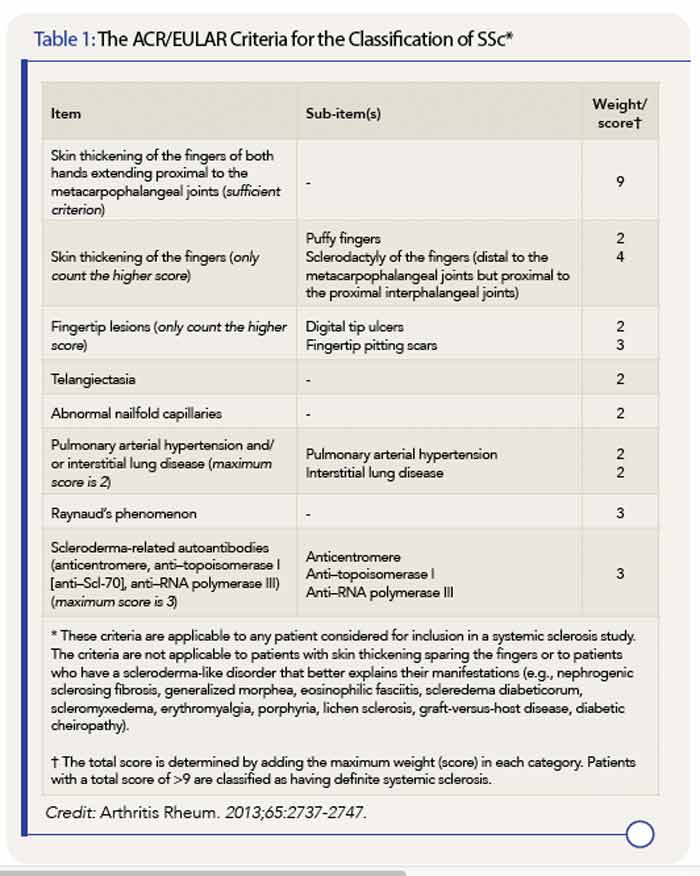
* These criteria are applicable to any patient considered for inclusion in a systemic sclerosis study. The criteria are not applicable to patients with skin thickening sparing the fingers or to patients who have a scleroderma-like disorder that better explains their manifestations (e.g., nephrogenic sclerosing fibrosis, generalized morphea, eosinophilic fasciitis, scleredema diabeticorum, scleromyxedema, erythromyalgia, porphyria, lichen sclerosis, graft-versus-host disease, diabetic cheiropathy).
† The total score is determined by adding the maximum weight (score) in each category. Patients with a total score of >9 are classified as having definite systemic sclerosis.
Credit: Arthritis Rheum. 2013;65:2737-2747.
Key Points in the New Criteria
The new classification criteria are considered superior to the 1980 criteria, with greater sensitivity and specificity. Most importantly, the classification system includes the three “hallmarks” of SSc—fibrosis of the skin and/or internal organs, production of certain autoantibodies, and vasculopathy.
Skin thickening of the fingers of both hands extending proximal to the metacarpophalangeal joints is the one criterion that, if present, can be used alone to classify a patient as having SSc. If that feature is not present, the point system created in the new criteria should be used. A score of nine or above, out of a possible 19 points, indicates a confirmed classification of SSc. Patients with skin thickening sparing the fingers are not, however, classified as having SSc.
Items assigned points in the scoring system are:
- Skin thickening of the fingers;
- Fingertip lesions;
- Telangiectasia;
- Abnormal nailfold capillaries;
- Pulmonary arterial hypertension and/or interstitial lung disease;
- Raynaud’s phenomenon; and
- Scleroderma-related autoantibodies (antitopoisomerase I, anticentromere, anti–RNA polymerase III).
The scoring system (see Table 1), kept to a maximum of 19 possible points with weights confined to single digits, should simplify its use, particularly given that similar systems are used for other rheumatic diseases, such as rheumatoid arthritis. Dr. Pope anticipates the creation of application software that will make the scoring system easier to use when classifying patients with SSc.
According to Dr. Pope, clinicians may wonder why certain items were not included in the list of classification criteria. Many were excluded because they were considered redundant, she says. For example, scleroderma renal crisis was excluded. “It is very specific but rare, so patients are usually classified by other features,” she says.
Some gastrointestinal features, such as lower esophageal dysphagia, dilated esophagus, and gastric antral vascular extasia, which are common in SSc and somewhat specific, were also considered redundant, “as other features are usually present to include these patients in the classification,” she says.
Cost of potential treatment and effect of the new criteria on insurance coverage were not part of the ACR/EULAR decision making or research in drafting this document. Dr. Pope noted that more patients potentially will be correctly classified with SSc and get access to medications that are indicated for certain organ system involvement. “There will be more overlap in the new criteria with clinical judgment where, previously, some patients were labeled with SSc but did not meet the former criteria.”
Kathy Holliman is a medical journalist based in New Jersey.
References
- Ringold S, Weiss PF, Beukelman T, et al. 2013 update of the 2011 American College of Rheumatology recommendations for the treatment of juvenile idiopathic arthritis: Recommendations for the medical treatment of children with systemic juvenile idiopathic arthritis and tuberculosis screening among children receiving biologic medications. Arthritis Care Res. 2013;65: 1551-1563.
- van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2013;65:2737-2747.
