Shidlovski/shutterstock.com
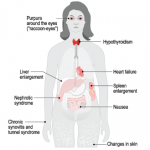
CHICAGO—Caryn A. Libbey, MD, clinical associate professor of medicine at Boston University School of Medicine, described the evolving in our understanding of amyloid at the ACR’s State-of-the-Art Clinical Symposium in April. Amyloidosis is a rare disease that is often underdiagnosed and undertreated.
“Even though this disease has been around for 150 years, I still consider it a relatively modern disease, because the first fibril was identified only in 1971,” explained Dr. Libbey. There are more than 30 amyloid precursor proteins, which, in healthy individuals, remain soluble. When proteins, such as the kappa or lambda light chain or transthyretin, become insoluble, they can cause amyloidosis.