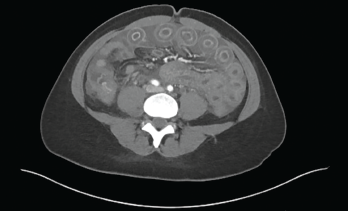
Figure 1: Computed Tomography of the Abdomen
Our patient was a 33-year-old, 5’2″ Asian woman with a past medical history of systemic lupus erythematosus (SLE). The diagnosis was based on serologies positive for anti-nuclear antibodies (ANAs), as well as antibodies to Sm, RNP and SSA. Her illness included neuropsychiatric and cutaneous involvement. She also had a diagnosis of Hashimoto’s thyroiditis.
She presented with two weeks of abdominal bloating and one week of abdominal pain, nausea, vomiting and non-bloody diarrhea. Her symptoms began two weeks before, with abdominal fullness and bloating, which progressed to acute-onset, diffuse, intermittent, cramping abdominal pain associated with non-bloody emesis and multiple episodes of non-bloody, watery diarrhea. She had not been able to tolerate oral intake for the past few days. She denied fevers, chills, sick contacts or recent travel, but said a sweet potato casserole she ate may have been “bad.”
On further questioning, she reported a similar episode occurred about 11 months before. That episode lasted for a few days and resolved after she was administered intravenous fluids at student health.
Lupus History
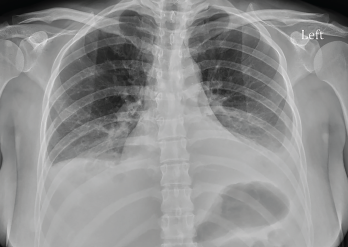
Figure 2: Radiograph of the Chest
Our patient was first diagnosed with SLE in 2014. She had acute cutaneous lupus, with biopsy-proven interface dermatitis (i.e., a pattern of skin reaction characterized by an inflammatory infiltrate that appears to obscure the dermo-epidermal junction when observed at low-power examination and referred to as lichenoid tissue reaction) and was initiated on 200 mg of hydroxychloroquine twice a day.
In 2015, she developed neuropsychiatric symptoms—seizures with an elevated immunoglobulin G (IgG) index on her cerebrospinal fluid analysis—as well as oral ulcers and alopecia. Serologies at that time were remarkable for an ANA of 1:1280 in a speckled pattern, anti-Sm, anti-RNP and anti-SSA antibodies, low complements and leukopenia.
She was treated with 1,000 mg of intravenous methylprednisolone daily for three days. Her treatment also included 1,500 mg of mycophenolate mofetil twice a day, 200 mg of hydroxychloroquine twice a day and a slow taper of prednisone.
She self-discontinued all of her medications toward the later part of 2015 due to what she described as medication side effects and a concern for toxicity.

