The PALACE study demonstrates a non-significant, 0.6 and 0.0 exposure-adjusted incidence rate/100 patient-years of opportunistic infection in placebo vs. apremilast, respectively.3 The ACTIVE trial produced consistent safety profile results.4 Even though no increased risk of opportunistic infections was identified, alterations in inflammatory cytokine levels were observed. This alteration of the inflammatory milieu may have immunosuppressive effects (see Figures 1 & 2, & 3).
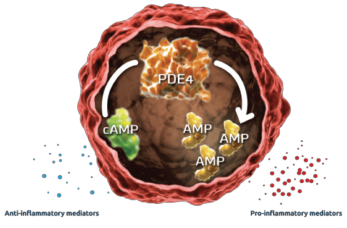
Figure 1: Mechanism of PDE45
Based on the PALACE study, the apremilast package insert does not mention an increased risk of opportunistic infection associated with the use of this drug.
In PsA, activated T cells and macrophages induce production of inflammatory cytokines and chemokines. The key cytokines in inducing the clinical features of psoriatic arthritis are IL-17, TNFα and IL-12/23. IL-17A is a proinflammatory cytokine produced by T helper 17 cells, macrophages, mast cells, dendritic cells, natural killer (NK) cells and CD8 T cells.
IL-17 is a key cytokine in the pathogenesis of PsA. Synovial biopsies from patients with PsA demonstrate higher levels of IL-17 than similar biopsies from patients with rheumatoid arthritis (RA).6 IL-17 works in targeted tissues including the keratinocyte (leading to epidermotropism of T cells and neutrophils and a subsequent psoriatic plaque), synovial fibroblast (with proliferation of the synovial fibroblast and induction of metalloproteinases that break down bone), endothelial cell (inducing angiogenesis and, paired with the activated synovial fibroblast, leading to inflammatory pannus of the synovium) and osteoclast (activating the RANK system and leading to osteoclastogenesis and formation of bone erosions).7
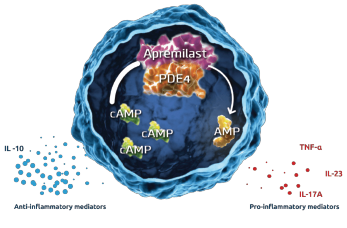
Figure 2: Inhibition of PDE4 by Apremilast5
TNFα is a proinflammatory cytokine overexpressed in the synovium, leading to synovial hyperproliferation, increased T cell and macrophage infiltration, induction of angiogenesis, osteoclast activation, synovial proliferation and metalloprotease release leading to joint destruction.8
IL-12 is produced by monocytes and macrophages in response to inflammation. Its activation results in stimulation and differentiation of Th1 cells, which leads to increased macrophages and triggering of a cellular immunity response.
IL-23 induces Th17 differentiation and subsequent increase of IL-17 and IL-22, producing subsequent downstream effects (including activation and inflammatory reaction synovio-entheseal complex and resulting in collagen enthesitis.)6
IL-22 acts on keratinocytes and synoviocytes leading to hyperkeratosis and dermal, epidermal and synovial inflammation (see Figure 4).