MOLEKUUL / Science Source
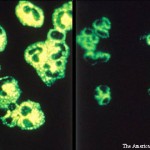
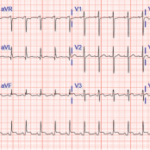
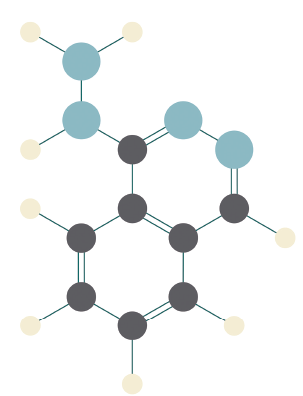
Drug-induced lupus erythematosus and ANCA-associated vasculitis (AAV) are both autoimmune conditions associated with the use of hydralazine, a commonly prescribed drug for hypertension and congestive heart failure. Although the pathogenesis is unknown, it is believed that hydralazine alters neutrophil and lymphocyte function and promotes exposure of antigens, leading to the development of anti-neutrophil antibodies (ANCA) and/or anti-nuclear antibodies (ANA) and ultimately triggering a systemic response.1
These conditions are rare and typically considered to be mutually exclusive. Here, we describe a rare presentation in which hydralazine appears to be the inciting factor of a patient developing an overlap of drug-induced lupus erythematosus and AAV.