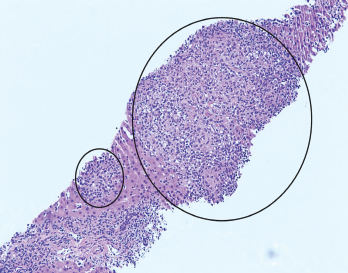
Figure 1. H&E stain at 100X showing granulomas accompanied by scattered admixed lymphocytes.
A 26-year-old woman presented to our emergency department (ED) with intermittent fevers, nausea and vomiting. She had a past medical history of well-controlled, anti-nuclear antibody positive and rheumatoid factor negative polyarticular juvenile idiopathic arthritis (pJIA) and Crohn’s disease. Her maintenance treatment consisted of monthly intravenous infliximab, 10 mg of oral methotrexate weekly and 20 mg of oral leflunomide daily.
The patient had been evaluated two weeks before at an ED in Chicago for sinus congestion and generalized malaise. She reported that a computed tomography (CT) scan of the chest showed “early pneumonia.” She was discharged home with a prescription for azithromycin for a lower respiratory tract infection and told to abstain from her immunosuppressive agents.
The patient was given a dose of acetaminophen before the lab results were available, and her fever and tachycardia resolved. Further evaluation in the ED revealed no reactive heterophile antibodies (i.e., monospot test), normal lipase and a PCR test for influenza that was negative. The patient declined admission for further evaluation despite physician recommendations but agreed to return if her symptoms worsened. She was encouraged to keep an outpatient appointment with her primary rheumatologist scheduled for four days later.
The patient returned to the ED two days later with persistent intermittent fevers, nausea and vomiting. She had no headache, and she denied new medications (other than azithromycin), recent medication changes, recreational drug use, over-the-counter supplements, incorrect dosing of current medications or travel outside the U.S. She had been taking methotrexate and leflunomide for 25 and 12 years, respectively. Infliximab infusions had been initiated two years before. Her pJIA and Crohn’s disease were asymptomatic on this treatment regimen, though she had not taken immunosuppressives since onset of the current illness.
On examination, her temperature was 38.5°C (101.3°F), her weight was 74 kg (163 lbs.), and her heart rate was 114 beats per minute. Her general exam was otherwise unremarkable, without focal deficits. Laboratory testing revealed a rising AST of 783 U/L and ALT of 650 U/L, total bilirubin of 1.6 mg/dL (RR: 0.2–1.3 mg/dL), alkaline phosphatase of 195 U/L (RR: 35–104 U/L), platelets of 127,000/mcL (RR: 145,000–445,000 mcL), C-reactive protein of 10.5 mg/dL (RR: <0.5 mg/dL) and erythrocyte sedimentation rate (ESR) of 92 mm/hr (RR: 0–25 mm/hr).
The initial evaluation focused on addressing potential causes of hepatocellular disease. Tests for cytomegalovirus, herpes simplex, Epstein-Barr virus, parvovirus, hepatitis A, B and C, HIV, anti-mitochondrial antibody, anti-smooth muscle antibody, serum copper, ceruloplasmin, alpha-1 antitrypsin level and acetaminophen level were unremarkable.
Ultrasound of her abdomen showed splenomegaly and prominence of periportal fat for which a broad differential was considered, including infectious, medication, metabolic and inflammatory causes. Initial blood cultures demonstrated no growth.
A hepatologist ordered tests for tick-borne diseases and further imaging. Computed tomography (CT) of the abdomen and pelvis showed nonspecific changes of the liver suggestive of hepatitis and partially visualized hilar lymphadenopathy. A liver biopsy was recommended to further evaluate worsening transaminitis.
On the second day of admission, the patient remained febrile. Laboratory tests showed ferritin of 11,806 ng/mL (RR: 6.2–137 ng/mL), a declining absolute neutrophil count of 0.88 k/mcL (RR: 2.0–9.3 k/mcL), platelets of 101 k/mcL, ESR of 25 mm/hr and triglycerides of 298 mg/dL (RR: <150 mg/dL), raising concern for macrophage activation syndrome (MAS).
Intravenous methylprednisolone, 1 g daily, was initiated, with the subsequent addition of 100 mg of subcutaneous anakinra every eight hours.
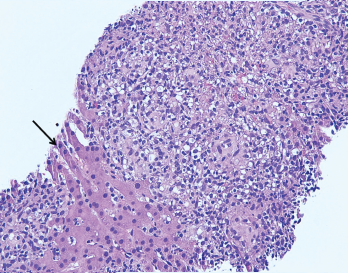
Figure 2. The image from Figure 1 at 200X showing granulomas accompanied by scattered admixed lymphocytes without discernible atypia. Adjacent hepatocytes (black arrow), polygonal cells with centrally placed nucleus and eosinophilic cytoplasm, can also be identified.
A liver biopsy performed on hospital day 3 revealed granulomatous hepatitis (see Figures 1 & 2) but no hemophagocytosis. Iron stains, periodic acid-Schiff diastase stain, immunohistochemistry for cytomegalovirus, and in situ hybridization for Epstein-Barr virus RNA were negative. Subsequent Grocott’s methenamine silver (GMS) stain showed scattered yeast forms (see Figure 3), morphologically consistent with Histoplasma capsulatum. Acid-fast bacillus and Gram stains were negative. Liver tissue culture was not obtained. Serum and urine histoplasma antigens and 1,3 beta-D-glucan were elevated.
A repeat CT of her chest with contrast demonstrated left lower lobe consolidation and incidentally revealed multiple hypodense splenic lesions attributed to a suspected underlying infectious process.
In the setting of multi-organ involvement of histoplasmosis (lung, liver, spleen and lymph nodes), treatment with intravenous liposomal amphotericin B (3 mg/kg/day) was initiated and methylprednisolone discontinued. Anakinra was continued to treat MAS secondary to disseminated fungal infection.
Bone marrow biopsy ultimately revealed hemophagocytosis with engulfment of erythrocyte precursors by macrophages, as well as scattered foci of loosely aggregated histiocytes suggestive of poorly formed granulomas. GMS stain was negative; bone marrow cultures were not performed. An interleukin-2 (IL-2) soluble receptor α was elevated at 8,600 U/mL (RR: 223–710 U/mL), further supporting a diagnosis of MAS.
The patient became consistently afebrile on hospital day 7, and lab abnormalities improved. On hospital day 12, she was discharged on anakinra at a dosage of 100 mg twice daily and a two-week course of intravenous liposomal amphotericin B. She remained off immunosuppressant medications.
Following discharge, the patient continued to improve. One month following hospital discharge, her laboratory abnormalities returned to normal. Upon completion of amphotericin, she transitioned to treatment with oral itraconazole. The serum and urine histoplasma antigen levels returned to normal after three months of itraconazole. She developed a pJIA flare while on anakinra without other immunosuppressive treatment. After discussion with consultants and her pediatric rheumatologist, she resumed infliximab with subsequent improvement.
The patient continues on oral itraconazole as a secondary histoplasma prophylaxis as long as she requires immunosuppressive therapy.
Discussion
Macrophage activation syndrome has been reported in most rheumatic diseases, typically triggered by infection (as seen in our patient) in the setting of immunosuppressive therapy. The syndrome is commonly described in association with systemic juvenile idiopathic arthritis (sJIA), in which viral infections remain the most commonly identified trigger, although a broad range of bacterial and fungal infections is capable of initiating the systemic inflammatory response. As seen here, histoplasmosis has been described as a trigger, but is noted only in case reports or case series.1
Diagnosis
The most common clinical features of MAS are high, non-remitting fever, hepatosplenomegaly, generalized lymphadenopathy, central nervous system dysfunction and hematologic abnormalities. Pyrexia is a cardinal feature of the disease.2 Hyperferritinemia is highly characteristic of MAS; a serum ferritin level >10,000 mg/L has a sensitivity and specificity of 96% and 90%, respectively.3 On the other hand, a low fibrinogen as a consequence of the hyperinflammatory state can result in a paradoxical drop in sedimentation rate.4
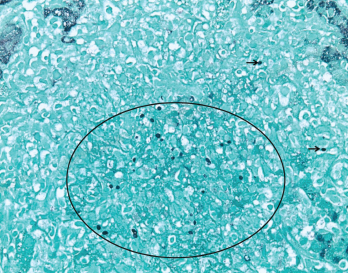
Figure 3. GMS (Grocott’s methenamine silver stain) at 400X highlights scattered yeast forms (black circle and black arrows) morphologically consistent with Histoplasma capsulatum.
The 2016 MAS classification criteria approved by the European League Against Rheumatism (EULAR) and the ACR call for a febrile patient with known sJIA to have a ferritin of >684 ng/mL as well as two other laboratory findings that may include a platelet count of 181,000/mL, AST >48 units/L, triglycerides >155 mg/dL and/or a fibrinogen ≤360 mg/dL.2 Although these criteria are helpful in the recognition of MAS, they were created to standardize the definition of the MAS patient for clinical studies. The condition remains difficult to definitively diagnose early in the disease course.
The hemophagocytic lymphohistiocytosis (HLH) 2004 guidelines are often used in clinical practice to aid diagnosis. One retrospective study assessed the ability of these criteria to differentiate between sJIA-associated MAS, active sJIA and systemic infectious diseases. When three out of five HLH-2004 criteria (i.e., fever, splenomegaly, peripheral blood cytopenias affecting two of three cell lineages, hypertriglyceridemia or hypofibrinogenemia, and evidence of hemophagocytosis) were met, these criteria reached 98% specificity in identifying sJIA patients with MAS vs. those with a systemic infectious disease (κ coefficient=0.74).5
The adapted HLH-2004 criteria did not include soluble interleukin-2 receptor (sIL-2r) because it was not collected in all studied patients. However, this marker remains a useful tool due to its low cost and commercial availability. The receptor is upregulated in activated T cells and thus found in high levels in hemophagocytic conditions, lymphomas and other conditions with T cell activation.
One single-center, retrospective study assessed the diagnostic value of this marker in 78 patients with suspected HLH (38 would be diagnosed using HLH-2004 criteria and 40 would have other diagnoses). An sIL-2r value of >2,515 U/mL yielded a test sensitivity of 100% and specificity of 72.5%, whereas a value of >10,000 U/mL increased the specificity to 93%.6
Hemophagocytosis does not correlate with MAS activity or clinical disease, and its absence should not exclude the diagnosis. It is recognized now that hemophagocytosis occurs later in the disease course.7 Our patient met all five of the adapted criteria, displayed an elevated sIL-2r, and her bone marrow biopsy revealed hemophagocytosis, collectively underscoring an MAS diagnosis and the need to treat both MAS and disseminated histoplasmosis. She improved with parallel therapy directed at both conditions.
Treatment
Successful treatment with anakinra in pediatric patients with MAS has been documented in multiple case reports over the years (doses 1 mg/kg/day to 2 mg/kg/day), and this immunosuppressive is used increasingly as an initial therapy.8
To better understand the role of anakinra in patients with features of MAS, a re-analysis of a prospective, randomized, double blind, placebo-controlled phase 3 trial evaluating all-cause mortality with addition of anakinra in adult patients with severe sepsis was performed on a smaller subset of patients with features of MAS, such as hepatobiliary dysfunction and disseminated intravascular coagulation. They were compared against those who had one or neither of these characteristics and actually showed an improvement in 28-day mortality.9 In this trial, a dose of 2 mg/kg/hr intravenously for 72 hours was studied.
No validated treatment protocols exist for MAS. Management strategies are derived from retrospective studies and from experience treating familial hemophagocytic lymphohistiocytosis. These approaches usually involve a combination of immunosuppressive agents. Although monotherapy with high-dose steroids has been described as successful, up to 50% of reported adult cases are resistant.10
Recent collaborative work has underscored the need for early and aggressive immunosuppression and simultaneous treatment targeting the underlying inciting infection. Other protocols call for pulse-dose glucocorticoids (i.e., 1 g of methylprednisolone daily for three to five days), followed by intravenous immunoglobulin 1 g/kg daily for two days, with anakinra should there be any clinical deterioration.10 Alternatively, anakinra in a 100 mg subcutaneous dose four times a day may be effective.11 Underlying disorders that may be contributing should be treated concurrently, and malignancy should be considered if a clear insult is not found. We chose the dose of 100 mg subcutaneously every eight hours (3.9 mg/kg/day) due to the efficacy and safety at higher doses alluded to in these trials. IL-1 inhibition is generally not associated with the risk of opportunistic infection.11
Disseminated histoplasmosis in immunocompromised patients may mimic MAS at onset. It also presents as an acute febrile illness associated with fatigue and weight loss.12 Histoplasmosis should be on the differential in immunocompromised patients with systemic febrile diseases, particularly those residing in endemic areas, such as the Midwestern and Central U.S., along the Ohio and Mississippi River valleys, as seen in our patient.
The estimated U.S. incidence of histoplasmosis in patients on infliximab is 18.78 per 100,000.13 National Medicare claims data suggests an incidence of 6.1 per 100,000 in the general population in the Midwestern U.S.14
Identification of recent exposure is difficult, and reactivation of latent histoplasmosis can occur years after initial exposure.15 Pulmonary symptoms, such as cough and dyspnea, may not be present. Hepatosplenomegaly and lymphadenopathy may be appreciated on physical exam or imaging. Laboratory testing commonly reveals pancytopenia and hepatic enzyme abnormalities, as may be seen in MAS. Some patients may present with or progress to severe shock with respiratory distress and multisystem organ failure.12
Central nervous system (CNS) involvement is present in 5–10% of cases of disseminated histoplasmosis and can present with a wide variety of clinical and imaging findings.16 Fortunately, our patient had no neurologic symptoms or exam abnormalities over her disease course, thus no lumbar puncture or neurologic imaging was obtained.
Establishing the diagnosis of disseminated histoplasmosis is challenging because malignancy, infection and other inflammatory conditions may present with similar findings. In these patients it is prudent to pursue the diagnosis through histopathology from affected sites and/or antigen detection.17 Cultures may take several weeks of incubation for a result.
Treatment recommendations from the Infectious Diseases Society of America in 2007 are based on severity of infection and the presence of CNS involvement.18 For moderate to severe disseminated disease, the recommended treatment is liposomal amphotericin B for one to two weeks, followed by oral itraconazole for a total of at least 12 months. Patients with CNS involvement should receive a longer course of liposomal amphotericin B at higher doses followed by oral itraconazole. All patients are advised to undergo routine serum and urine antigen testing to assess response to treatment. Some patients with underlying immunocompromising conditions may benefit from lifelong suppressive therapy with itraconazole.
In Sum
We present a case of a young female from the Midwest on immunosuppressive treatment for pJIA and Crohn’s disease who was evaluated for fevers, transaminitis, hyperferritinemia and an acute inflammatory response. The evaluation revealed disseminated histoplasmosis complicated by MAS. In this setting, consideration should be given to both infectious triggers of MAS and MAS mimickers, of which disseminated histoplasmosis remains a rare but important type.
 Osman Bhatty, MD, is currently a practicing rheumatologist at Advocate Aurora Health, Chicago.
Osman Bhatty, MD, is currently a practicing rheumatologist at Advocate Aurora Health, Chicago.
 Dale Kobrin, MD, is an internal medicine resident at Allegheny General Hospital, Pittsburgh, Pa., and will be starting rheumatology fellowship at the National Institutes of Health in July 2022.
Dale Kobrin, MD, is an internal medicine resident at Allegheny General Hospital, Pittsburgh, Pa., and will be starting rheumatology fellowship at the National Institutes of Health in July 2022.
 Lauren Mathos, DO, is an academic internist practicing primary care at Allegheny Health Network, Pittsburgh, Pa.
Lauren Mathos, DO, is an academic internist practicing primary care at Allegheny Health Network, Pittsburgh, Pa.
Nazia Khatoon, MD, completed her pathology training at Allegheny Health Network.
 Yazan Samhouri, MD, is a hematologist and cellular therapist. He is in practice at Allegheny Health Network Cancer Institute, Pittsburgh, Pa.
Yazan Samhouri, MD, is a hematologist and cellular therapist. He is in practice at Allegheny Health Network Cancer Institute, Pittsburgh, Pa.
 Naga Sai Krishna Patibandla, MD, is a third-year hematology oncology fellow at Allegheny Health Network, Pittsburgh, Pa.
Naga Sai Krishna Patibandla, MD, is a third-year hematology oncology fellow at Allegheny Health Network, Pittsburgh, Pa.
 Mary Chester M. Wasko, MD, MSc, is emeritus professor of rheumatology for the Allegheny Health Network, Pittsburgh, Pa.
Mary Chester M. Wasko, MD, MSc, is emeritus professor of rheumatology for the Allegheny Health Network, Pittsburgh, Pa.
References
- Agarwal S, Moodley J, Ajani Goel G, et al. A rare trigger for macrophage activation syndrome. Rheumatol Int. 2011 Mar;31(3):405–407.
- Ravelli A, Minoia F, Davì S, et al. 2016 classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: A European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation collaborative initiative Arthritis Rheumatol. 2016 Mar;68(3):566–576.
- Allen CE, Yu X, Kozinetz C, et al. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008 Jun;50(6):1227–1235.
- Lerkvaleekul B, Vilaiyuk S. Macrophage activation syndrome: Early diagnosis is key. Open Access Rheumatol. 2018 Aug 31;10:117–128.
- Davì S, Minoia F, Pistorio A, et al. Performance of current guidelines for diagnosis of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Arthritis Rheumatol. 2014 Oct;66(10):2871–2880.
- Hayden A, Lin M, Park S, et al. Soluble interleukin-2 receptor is a sensitive diagnostic test in adult HLH. Blood Adv. 2017 Dec;1(26):2529–2534.
- C, Yao X, Tian L, et al. Marrow assessment for hemophagocytic lymphohistiocytosis demonstrates poor correlation with disease probability. Am J Clin Pathol. 2014 Jan;141(1):62–71.
- Henderson LA, Cron RQ. Macrophage activation syndrome and secondary hemophagocytic lymphohistiocytosis in childhood inflammatory disorders: Diagnosis and management. Paediatr Drugs. 2020 Feb;22(1):29–44.
- Shakoory B, Carcillo JA, Chatham WW, et al. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: Reanalysis of a prior phase III trial. Crit Care Med. 2016 Feb;44(2):275–281.
- Carter SJ, Tattersall RS, Ramanan AV. Macrophage activation syndrome in adults: Recent advances in pathophysiology, diagnosis and treatment. Rheumatology (Oxford). 2019 Jan 1;58(1):5–17.
- Salvana EM, Salata RA. Infectious complications associated with monoclonal antibodies and related small molecules. Clin Microbiol Rev. 2009 Apr;22(2):274–290.
- Wheat LJ. Diagnosis and management of histoplasmosis. Eur J Clin Microbiol Infect Dis. 1989 May;8(5):480–490.
- Wallis RS, Broder M, Wong J, et al. Reactivation of latent granulomatous infections by infliximab. Clin Infect Dis. 2005 Aug 1;41 Suppl 3:S194–S198.
- Baddley JW, Winthrop KL, Patkar NM, et al. Geographic distribution of endemic fungal infections among older persons, United States. Emerg Infect Dis. 2011 Sep;17(9):1664–1669.
- Keath EJ, Kobayashi GS, Medoff G. Typing of Histoplasma capsulatum by restriction fragment length polymorphisms in a nuclear gene. J Clin Microbiol. 1992 Aug;30(8):2104–2107.
- Wheat J, Myint T, Guo Y, et al. Central nervous system histoplasmosis: Multicenter retrospective study on clinical features, diagnostic approach and outcome of treatment. Medicine (Baltimore). 2018 Mar;97(13):e0245.
- Kauffman CA. Histoplasmosis: A clinical and laboratory update. Clin Microbiol Rev. 2007 Jan;20(1):115–132.
- Wheat LJ, Freifeld AG, Kleiman MB, et al. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis. 2007 Oct;45(7):807–825.