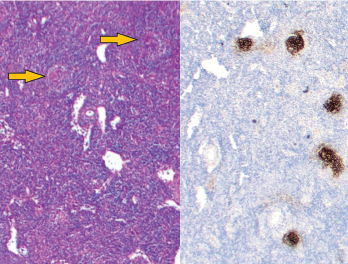
Figure 1: Vague nodular areas (orange arrows) on the left and CD21 staining for follicular dendritic cells on the right demonstrate follicles with regressed germinal centers.
The difference between Castleman disease and Castleman-like disease may be subtle, but it comes with significant ramifications.
Case Presentation
This case involves a pregnant 19-year-old woman who presents over multiple hospitalizations with concerns for systemic lupus erythematosus and macrophage activation syndrome.
At 36 weeks’ gestation, the patient’s weight had dropped from 215 lbs. to 170 lbs., and she was observed to have a hyperpigmented macular rash on her trunk and extensor surface of the arms.
Laboratory values at her obstetrics clinic visit were notable for elevated total bilirubin of 3.0 mg/dL (reference range [RR]: 0.4–1.5 mg/dL), aspartate aminotransferase (AST) 592 U/L (RR: 15–40 U/L), alanine aminotransferase (ALT) 245 U/L (RR: 14–50 U/L), alkaline phosphatase 155 U/L (RR: 24–110 U/L) and bile acids 107 umol/L (RR: 3.7–14.5 umol/L).
She was admitted for further examination and testing, at which time she was noted to have elevated inflammatory markers with an erythrocyte sedimentation rate (ESR) of 65 mm/hour (RR: 0–15 mm/hr) and a C-reactive protein (CRP) of 2.8 mg/dL (RR: <0.60 mg/dL). Her ANA was 1:2560 titer in a homogeneous pattern, and complement levels were low: C3: 74 mg/dL (RR: 86–208 mg/dL); and C4: 12 mg/dL (RR: 13–47 mg/dL). Anti-Ro, anti-La, anti-Smith, anti-RNP, anti-dsDNA, anti-smooth muscle and anti-mitochondrial antibodies were negative.
Due to her significantly elevated bile acids, she was given betamethasone intramuscularly to accelerate fetal lung maturity, and underwent emergent Cesarean section. Her symptoms and laboratory values quickly improved, and she was discharged.
Four months later, she presented to the emergency department with abdominal pain, joint pain and a temperature of 103°F, at which time she was noted to have altered mentation with slow, circular speech. Her laboratory values were significant for hemoglobin of 7 g/dL (RR: 12–15 g/dL), haptoglobin 4 mg/dL (RR: 30–200 mg/dL), direct Coombs’ with IgG 3+ (normal=negative), normal creatinine, urinalysis with trace blood and a 24-hour urine protein of 2,200 mg/g (RR: <100 mg/g). Her inflammatory markers were markedly elevated, with ESR of 117 mm/hour (RR: 0–15 mm/hr), CRP 9.8 mg/dL (RR: <0.60 mg/dL) and ferritin greater than 15,000 ng/mL (RR: 10–200 ng/mL).
Cerebral spinal fluid (CSF) studies were noninflammatory with two nucleated cells x106/L (RR: 0–5×106/L), protein 11 mg/dL (RR: 15–50 mg/dL) and glucose 76 mg/dL. There were no CSF oligoclonal bands (normal: <4 bands) and infectious studies were negative.
Given the patient’s hyperferritinemia, there was concern for macrophage activation syndrome secondary to systemic lupus erythematosus (SLE).
She was treated with dexamethasone and subcutaneous anakinra with rapid improvement of abdominal pain, joint pain and mental status. Despite being drawn two days after immunosuppression was initiated, the soluble interleukin 2 receptor (sIL-2R) was elevated at 2,200 U/mL (RR: <1,100 U/mL. NK cell function was normal.
Given her degree of proteinuria, a renal biopsy was performed. It showed podocyte hypertrophy and effacement, without evidence of immune-complex-mediated disease, focal segmental glomerulosclerosis or thrombotic microangiopathy. Given the possibility of atypical renal involvement from SLE, she was prescribed mycophenolate mofetil and prednisone (of note, patient’s baby was formula fed and not breastfed). Unfortunately, the patient did not follow up as an outpatient, and it was unclear whether she took the prescribed medications.
Two months later, the patient was found unresponsive at home after a generalized tonic-clonic seizure and brought by paramedics to the emergency department, with a temperature of 106°F, hypotensive and tachycardic. She was intubated, placed on a norepinephrine infusion and started on broad-spectrum empiric antibiotics. Other than the past medical history of SLE from recent months, she did not have known comorbidities.