The patient was treated with intravenous immunoglobulin, pulse steroids, hydroxychloroquine and mycophenolate. His skin lesions initially improved somewhat with treatment, only to worsen within three weeks. He was refractory to a second round of intravenous immunoglobulin. One month later, the patient developed multiple cutaneous tumors with a 12 lb. weight loss. A lesion biopsy confirmed mycosis fungoides (see Figures 1C–D). Despite radiation and chemotherapy, the patient died.
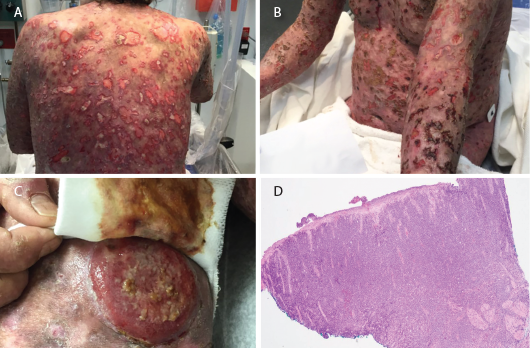
Figures 1 A–B: Numerous ulcerative lesions with sharply angulated edges are shown.
Figure 1C: A new tumorous lesion is shown on the patient’s leg.
Figure 1D: A biopsy of an ulcerated skin segment revealed an atypical mononuclear infiltrate.
Discussion
The association between malignancy and idiopathic inflammatory myopathy has long been reported. Overall, the malignancy risk in patients with myositis has been found to be 4.5-fold higher than in patients without idiopathic inflammatory myopathy. The relative risk of overall malignancy is 5.5 for DM and 1.62 for polymyositis. The relative risk was also significantly higher within the first year of diagnosis and higher in men.5
Proposed pathogenetic mechanisms of malignancy in autoimmune diseases include genetic predisposition, chronic immune stimulation and infections.6 Malignancy could also result from cross-reaction between muscle and tumor antigens, a paraneoplastic process or immunosuppressive treatments.7 DM often improves with treatment of malignancy, suggesting a parallel course.2
An analysis of the frequency and risk of specific cancers in idiopathic inflammatory myopathy found that of 618 DM patients, only three of 115 malignancies were non-Hodgkin’s lymphoma. Although the number of cases was low, the standardized incidence ratios were high. Ovarian and lung cancer had the highest standardized incidence ratios when compared with non-Hodgkin’s lymphoma.2
Skin is the second most frequently involved extranodal site for non-Hodgkin’s lymphoma, after the gastrointestinal tract. Most primary cutaneous non-Hodgkin’s lymphomas are T cell derived, but nodal non-Hodgkin’s lymphoma is B cell derived. Mycosis fungoides and Sézary syndrome are the two most common of 12 subtypes of cutaneous T cell lymphomas.8
In our literature review of DM, we found only six cases of cutaneous T cell lymphoma, with only one patient having mycosis fungoides. Despite its rarity, cutaneous T cell lymphoma should be considered in DM patients presenting with such skin findings as atypical plaques, nodules, ulcerations and tumor formation.3 Some of these findings were noted in our patient (see Figures 1A–D).
Because cutaneous T cell lymphoma lesions mimic those of other diseases and can be mistaken for atypical DM rash, diagnosis can be delayed. Sequential biopsies may be necessary to clinch the diagnosis and initiate proper treatment, as required in our patient (see Table 1).
Table 1: Description of Sequential Skin Biopsies
| Biopsy #1 | Interface dermatitis |
|---|---|
| Biopsy #2 | Dermal small vessel thrombotic vasculitis and coagulopathy with epidermal necrosis and fibrinopurulent inflammation (see Figures 1A–B) |
| Biopsy #3 | Lichenoid atypical large T cell lymphocytic infiltrate. Lymphocytes in the papillary dermis had cytologic atypia and strong positivity for CD3 and CD7. CD20 and CD30 were negative. |
| Biopsy #4 | Abnormal mononuclear infiltrate consistent with ulcerated T cell lymphoma/leukemia. Final diagnosis: Tumor stage mycosis fungoides, CD30 negative, large cell transformation. (see Figures 1C and 1D) |
Prognosis of mycosis fungoides depends on the extent of the involvement. Patients with limited skin involvement have a better prognosis; patients with diffuse skin involvement, tumor development or extracutaneous involvement have poor prognosis with increased mortality.9
Treatment options for mycosis fungoides in the advanced stage include chemotherapy, photodynamic therapy, radiation and bone marrow transplant.10