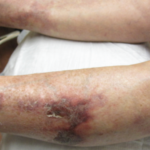
The most important aspect of treating patients with calciphylaxis remains wound management.
Discussion
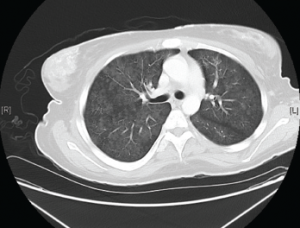
Figure 2. Diffuse bilateral groundglass opacities were seen throughout the lung parenchyma.
Background: Calciphylaxis is a cutaneous ischemic small-vessel vasculopathy that can lead to multiorgan calcification and necrosis. It’s most commonly seen in uremic patients with end-stage renal disease (ESRD) on dialysis. Approximately 1–4% of patients with ESRD develop calciphylaxis.1 It’s much rarer in non-uremic patients.2 A systematic review in 2008 found 36 reported cases of non-uremic calciphylaxis. Of those 36 cases, only four (11.1%) were related to connective tissue diseases.2 So far, only two reported cases of systemic lupus erythematosus developed non-uremic calciphylaxis.3,4
Pathogenesis: It remains largely unclear why certain people develop calciphylaxis. In patients with chronic kidney disease, it’s widely thought to be secondary to the disruption of calcium homeostasis due to secondary hyperparathyroidism.5
At the molecular level, some have proposed that vascular calcification is driven via downregulation of certain inhibitors of vascular calcifications. Of those, the ones implicated are the fetuin-A glycoproteins and matrix Gla proteins (MGP). Fetuin-A is a glycoprotein that binds calcium and phosphate and clears excess calcium phosphate from circulation, thereby inhibiting vascular calcification. In cases of vitamin D deficiency, downregulation of fetuin-A has been observed. MGP is a mineral-binding extracellular matrix protein synthesized by vascular smooth muscle, endothelium and chondrocytes. It’s been shown to inhibit artery calcification in animal models.6
Other proposed mechanisms include the upregulation of NFκB activity via the RANK-RANKL pathway. Increased NFκB activity has been shown to cause vascular mineral deposition.7
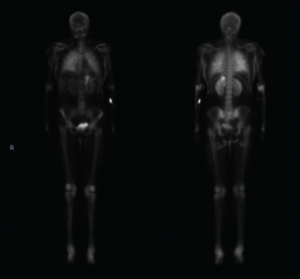
Figure 3. This nuclear bone scan shows uptake in bilateral lungs, soft tissues of the upper and lower extremities, and the stomach.
Unfortunately, none of these models explain calciphylaxis in patients without any evidence of calcium homeostasis imbalance.