crystal light / shutterstock.com
Calciphylaxis is a poorly understood and life-threatening ischemic vasculopathy characterized by calcification of the small- and medium-size arteries in the skin, subcutaneous tissue and internal organs, which leads to thrombosis, tissue necrosis and painful skin ulcerations that won’t heal. The disease has a 50–80% mortality rate. Although affected patients typically have end-stage renal disease (ESRD) and associated hyperparathyroidism, it can (rarely) occur in non-uremic patients. As of now, only a handful of reported cases of autoimmune disease patients with non-uremic calciphylaxis exist.
Below, we present the case of a 25-year-old female with systemic lupus erythematosus (SLE) and class I lupus nephritis with preserved renal function (GFR>100) who had biopsy-proved calciphylaxis. Despite comprehensive, multidisciplinary efforts and the use of numerous different treatment modalities over a three-month hospitalization period, the disease continued to progress, eventually leading to sepsis and death.
We hope this case raises awareness of this rare, fatal disease in the setting of an underlying autoimmune disease and provides a comprehensive overview of the disease, with an emphasis on the treatment modalities currently available.
In patients with chronic kidney disease, calciphylaxis is widely thought to be secondary to the disruption of calcium homeostasis due to secondary hyperparathyroidism.
The Case Report
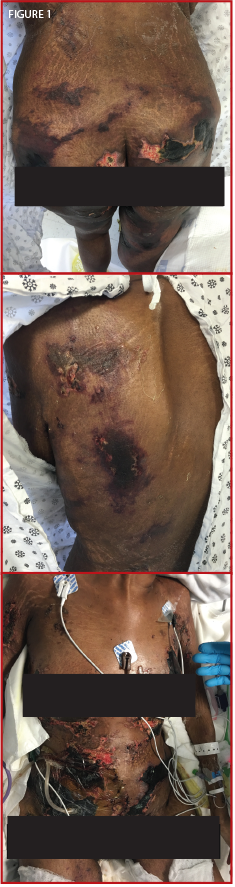
Figure 1. The patient presented confluent, hyperpigmented plaques over her thighs, buttocks, back, breasts and abdomen.
A 25-year-old black female with a two-year SLE history with features of arthritis, discoid skin lesions and class I lupus nephritis presented with a two-week history of hyperpigmented skin lesions on her arms, thighs, chest and back, associated with worsening pain. Her immunosuppressive regimen at the time consisted of mycophenolate mofetil 1,500 mg BID, hydroxychloroquine 400 mg QD, prednisone 5 mg QD and monthly belimumab infusions. Examination revealed confluent hyperpigmented plaques distributed across her upper extremities, thighs, chest and back (see Figure 1).
Laboratory data upon initial review were significant for profound anemia, with a hemoglobin of 6.3 g/dL and hematocrit of 21%. Her erythrocyte sedimentation rate (ESR) was elevated to 118 mm/h with a C-reactive protein (CRP) of 1.5 mg/dL; lactate dehydrogenase (LDH) and haptoglobin remained within normal limits. DsDNA was elevated at 193.9 IU, C4 was low at 5 mg/dL and C3 was normal at 94 mg/dL. However, when compared with her previous lupus serologies, the complements and dsDNA were close to her baseline. Otherwise, serum creatinine (0.8 mg/dL), calcium (10.3 mg/dL), phosphorus (3.7 mg/dL), glomerular filtration rate (GFR), liver function tests and the rest of her basic metabolic panel proved unremarkable (see Table 1).
Initially, given the elevated inflammatory markers, new skin lesions and low complements (i.e., C4), the patient was treated with prednisone 60 mg orally once daily for a possible lupus flare. Despite high steroid doses, the patient’s skin lesions continued to spread and worsen. A biopsy result of her skin lesions was obtained, showing diffuse calcium deposits throughout the dermis and subcutaneous layers (including the blood vessels), which was consistent with calciphylaxis.
An extensive workup was performed to determine the etiology of the patient’s calciphylaxis. It involved the evaluation for anti-phospholipid syndrome (APLS), cryoglobulinemia, anti-neutrophil cytoplasmic antibody (ANCA) associated vasculitis, endocarditis, septic emboli, malignancy and hyperparathyroidism. Unfortunately, this workup proved inconclusive. Parathyroid hormone (PTH), parathyroid hormone-related protein (PTHrP), cryoglobulin levels, myeloperoxidase antibodies (MPO), proteinase 3 antibodies (PR3), beta-2-glycoprotein, cardiolipin antibodies, lupus anticoagulant, creatinine kinase and serum protein electrophoresis (SPEP) were all negative or within normal limits.
A computed tomography (CT) scan of the chest, abdomen and pelvis revealed diffuse subcutaneous calcifications and diffuse bilateral groundglass opacities in the lungs (see Figure 2). There were no masses or nodules suspicious for malignancy. A nuclear bone scan demonstrated abnormal radiotracer uptake in bilateral lungs, soft tissues in the upper and lower extremities, and her stomach, which was consistent with calciphylaxis (see Figure 3).
Initially, the patient was started on sodium thiosulfate 25 g IV three times per week. Prednisone was tapered down quickly to 20 mg once a day to aid with wound healing and minimize infection risk. Both mycophenolate mofetil and belimumab were withheld during patient’s initial hospitalization to minimize further infection risk. Despite these efforts, the wounds rapidly worsened over the following week and a half.
Given the concern the patient’s underlying connective tissue disease (CTD) was driving the calciphylaxis, intravenous immunoglobulin therapy (IVIG) was given at a dose of 2 mg/kg split into three consecutive days. However, the patient’s skin lesions continued to spread. Sodium thiosulfate was discontinued after approximately two weeks of therapy, and the patient was given a trial of pamidronate 30 mg IV. Unfortunately, the patient became hypocalcemic and could not receive further pamidronate doses. The patient also received plasmapheresis every other day for a total of five sessions over a two-week period, during which the lesions appeared to stabilize, but then started getting worse again.
The patient was transferred to a nearby hospital for a trial of hyperbaric oxygen therapy, but was deemed not a candidate due to severe mitral regurgitation in the setting of a ruptured chordae tendonae as a result of calciphylaxis affecting the heart structures.
She was given a trial of intralesional sodium thiosulfate, but that proved ineffective as well. She continued to receive monthly IVIG therapy to manage her lupus, and prednisone was gradually tapered. But when prednisone was decreased below 15 mg, she developed synovitis, prompting re-uptitration of her prednisone as well as re-initiation of low-dose mycophenolate mofetil at 500 mg twice a day.
Throughout the course, the patient developed intermittent skin infections, requiring her immunosuppressants (i.e., mycophenolic acid or CellCept) to be discontinued. Eventually, the patient developed septic shock secondary to multiple nonhealing wound infections, leading to the deterioration of her health. She died after four months of struggling against a very aggressive and resistant calciphylaxis.
The most important aspect of treating patients with calciphylaxis remains wound management.
Discussion
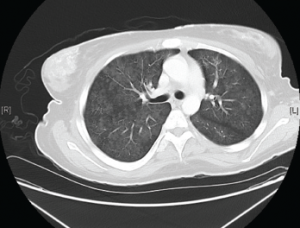
Figure 2. Diffuse bilateral groundglass opacities were seen throughout the lung parenchyma.
Background: Calciphylaxis is a cutaneous ischemic small-vessel vasculopathy that can lead to multiorgan calcification and necrosis. It’s most commonly seen in uremic patients with end-stage renal disease (ESRD) on dialysis. Approximately 1–4% of patients with ESRD develop calciphylaxis.1 It’s much rarer in non-uremic patients.2 A systematic review in 2008 found 36 reported cases of non-uremic calciphylaxis. Of those 36 cases, only four (11.1%) were related to connective tissue diseases.2 So far, only two reported cases of systemic lupus erythematosus developed non-uremic calciphylaxis.3,4
Pathogenesis: It remains largely unclear why certain people develop calciphylaxis. In patients with chronic kidney disease, it’s widely thought to be secondary to the disruption of calcium homeostasis due to secondary hyperparathyroidism.5
At the molecular level, some have proposed that vascular calcification is driven via downregulation of certain inhibitors of vascular calcifications. Of those, the ones implicated are the fetuin-A glycoproteins and matrix Gla proteins (MGP). Fetuin-A is a glycoprotein that binds calcium and phosphate and clears excess calcium phosphate from circulation, thereby inhibiting vascular calcification. In cases of vitamin D deficiency, downregulation of fetuin-A has been observed. MGP is a mineral-binding extracellular matrix protein synthesized by vascular smooth muscle, endothelium and chondrocytes. It’s been shown to inhibit artery calcification in animal models.6
Other proposed mechanisms include the upregulation of NFκB activity via the RANK-RANKL pathway. Increased NFκB activity has been shown to cause vascular mineral deposition.7
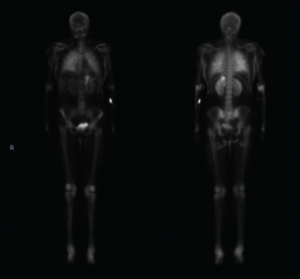
Figure 3. This nuclear bone scan shows uptake in bilateral lungs, soft tissues of the upper and lower extremities, and the stomach.
Unfortunately, none of these models explain calciphylaxis in patients without any evidence of calcium homeostasis imbalance.
Clinical Presentation & Diagnosis: Clinicians should suspect calciphylaxis when patients present with painful ischemic skin necrosis. In some cases, pain can precede skin lesions. Typical areas involved include the abdomen, thighs and buttocks. These areas typically feature an abundant amount of adipose tissue.1
Calciphylaxis is a clinical diagnosis. Although biopsies can help, they can worsen ulcerations and infections. Sometimes bone scans can help identify soft-tissue calcifications.8,9
Management: To adequately treat calciphylaxis, clinicians must perform an evaluation for reversible calciphylaxis causes. Workups should include checking calcium, phosphate and PTH levels. Also perform a thorough evaluation for malignancies and connective tissue diseases.10
The most widely used therapy is sodium thiosulfate. It’s thought that sodium thiosulfate works by removing calcium through chelation. In animal models, it’s been shown to transiently decrease ionized calcium and increase urinary calcium excretion.11
More than 51 cases of calciphylaxis have been successfully treated. Clinical response typically occurs within one or two weeks, with pain alleviation occurring first and wound healing following up to eight weeks after.12,13
Bisphosphonates have also been used to treat calciphylaxis. In six reported cases, either pamidronate or etidronate were used to treat uremic calciphylaxis. In two reported cases, bisphosphonates successfully treated patients with non-uremic calciphylaxis.14,15,16 Some case reports have shown responses as early as two days post-pamidronate infusion. The exact mechanism of how bisphosphonates reduce calciphylaxis remains unknown, but others have suggested bisphosphonates may inhibit macrophages and thereby suppress inflammatory cytokine release.17 Others have suggested the mechanism of action occurs via inhibition of calcium phosphate crystal formation, which has been implicated in ectopic calcifications.15
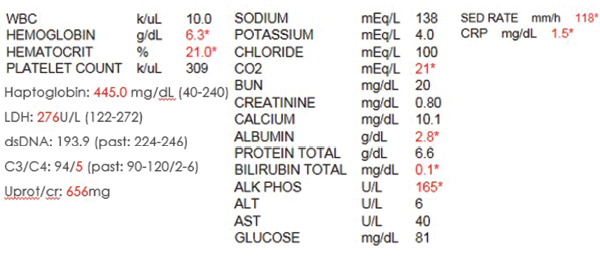
Table 1. The initial laboratory findings.
In patients with hyperparathyroidism, calcimimetics, such as cinacalcet, have shown much success in treating this rare, deadly illness. To date, nine cases of calciphylaxis have been successfully treated with calcimimetics used as an adjunctive therapy.18 Calcimimetics function by increasing calcium receptor sensitivity, thereby decreasing PTH levels.
But remember: Regardless of the above-mentioned treatments, the most important aspect of treating patients with calciphylaxis remains wound management. Due to the high mortality rate associated with the condition secondary to infections, having a wound-care specialist involved early on is crucial. In severe cases, surgical debridement may prove necessary. Hyperbaric oxygen therapy has also aided in wound healing. Research suggests hyperbaric oxygen stimulates fibroblast proliferation and neoangiogenesis, promoting arteriolar constriction to prevent tissue reperfusion injuries.19 High concentrations of oxygen are directly toxic to many infectious organisms, which helps prevent wound infections. A recent retrospective study of 46 cases showed 58% of the patients improved with hyperbaric oxygen therapy.20
Conclusion
Calciphylaxis remains poorly understood with poor outcomes. Cases of non-uremic calciphylaxis remain rare. Rarer still are reports of calciphylaxis in patients with an underlying connective tissue disease. Of the cases reviewed of patients with underlying SLE who had calciphylaxis, the majority ended in death due to severe infections.
Hopefully, this case report not only raises awareness of the condition, but helps clinicians facing similar challenges.
Joey Kim, MD, is a second-year rheumatology fellow at the Montefiore Medical Center in New York City.
Navneet Kaur, MD, is a second-year internal medicine resident at the Jacobi Medical Center in New York City.
Phillip Zhang, MD, is a third-year internal medicine resident at the Montefiore Medical Center in New York City.
Irene Blanco, MD, MS, is an associate professor of Medicine and the Rheumatology Fellowship Program director at the Albert Einstein College of Medicine in New York City.
References
- Janigan DT, Hirsch DJ, Klassen GA, et al. Calcified subcutaneous arterioles with infarcts of the subcutis and skin (“calciphylaxis”) in chronic renal failure. Am J Kidney Dis. 2000 Apr;35(4):588–597.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: A systematic review. Clin J Am Soc Nephrol. 2008 Jul;3(4):1139–1143.
- Lee JL, Naguwa SM, Cheema G, et al. Recognizing calcific uremic arteriolopathy in autoimmune disease: An emerging mimicker of vasculitis. Autoimmun Rev. 2008 Sep;7(8):638–643.
- Aliaga LG, Barreira JC. Calciphylaxis in a patient with systemic lupus erythematosus without renal insufficiency or hyperparathyroidism. Lupus. 2012 Mar;21(3):329–331.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): A complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995 Dec;33(6):954–962.
- Price PA, Omid N, Than TN, et al. The amino bisphosphonate ibandronate prevents calciphylaxis in the rat at doses that inhibit bone resorption. Calcif Tissue Int. 2002 Oct;71(4):356–363.
- Weenig RH. Pathogenesis of calciphylaxis: Hans Selye to nuclear factor kappa-B. J Am Acad Dermatol. 2008 Mar;58(3):458–471.
- Mochel MC, Arakaki RY, Wang G, et al. Cutaneous calciphylaxis: retrospective histopathologic evaluation. Am J Dermatopathol. 2013 Jul; 35(5):582–586.
- Paul S, Rabito CA, Vedak P, et al. The role of bone scintigraphy in the diagnosis of calciphylaxis. JAMA Dermatol. 2017 Jan 1;153(1):101–103.
- Yu Z, Gu L, Pang H, et al. Sodium thiosulfate: An emerging treatment for calciphylaxis in dialysis patients. Case Rep Nephrol Dial. 2015 Mar 13;5(1):77–82.
- Pasch A, Schaffner T, Huynh-Do U, et al. Sodium thiosulfate prevents vascular calcifications in uremic rats. Kidney Int. 2008 Dec;74(11):1444–1453.
- Nigwekar SU, Brunelli SM, Meade D, et al. Sodium thiosulfate therapy for calcific uremic arteriolopathy. Clin J Am Soc Nephrol. 2013 Jul;8(7):1162–1170.
- Hackett BC, McAleer MA, Sheehan G, et al. Calciphylaxis in a patient with normal renal function: Response to treatment with sodium thiosulfate. Clin Exp Dermatol. 2009 Jan;34(1):39–42.
- Monney P, Nguyen QV, Perroud H, et al. Rapid improvement of calciphylaxis after intravenous pamidronate therapy in a patient with chronic renal failure. Nephrol Dial Transplant. 2004 Aug;19(8):2130–2132.
- Shiraishi N, Kitamura K, Miyoshi T, et al. Successful treatment of a patient with severe calcific uremic arteriolopathy (calciphylaxis) by etidronate disodium. Am J Kidney Dis. 2006 Jul;48(1):151–154.
- Lorriaux A, Chaby G, Dhaille F, et al. Nonuraemic calciphylaxis: Response to treatment with pamidronate and negative pressure therapy. Clin Exp Dermatol. 2015 Jan;40(1):52–55.
- Hasegawa J, Nagashima M, Yamamoto M, et al. Bone resorption and inflammatory inhibition efficacy of intermittent cyclical etidronate therapy in rheumatoid arthritis. J Rheumatol. 2003 Mar;30(3):474–479.
- Sharma A, Burkitt-Wright E, Rustom R. Cinacalcet as an adjunct in the successful treatment of calciphylaxis. Br J Dermatol. 2006 Dec;155(6):1295–1297.
- Tibbles PM, Edelsberg JS. Hyperbaric-oxygen therapy. N Engl J Med. 1996 Jun 20;334(25):1642–1648.
- An J, Devaney B, Ooi KY, et al. Hyperbaric oxygen in the treatment of calciphylaxis: A case series and literature review. Nephrology (Carlton). 2015 Jul;20(7):444–450.