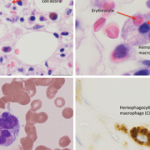
His repeat lab evaluation was notable for a markedly elevated ferritin of 29,930 ng/mL, elevated triglycerides at 210 mg/dL, anemia and thrombocytopenia, along with persistent transaminitis, coagulopathy and hyperammonemia. Liver and bone marrow biopsies demonstrated erythrophagocytosis consistent with HLH.
The patient’s immunologic evaluation was notable for hypogammaglobulinemia and neutropenia. Serum CD25 levels and natural killer (NK) cell functional studies (CD107a mobilization and perforin/granzyme B expression) were normal. He also had an extensive infectious evaluation of his blood, urine, stool, spinal fluid and respiratory secretions, none of which identified a pathogenic organism.
The patient was initially treated with ammonia-scavenger therapy and fresh frozen plasma (FFP) for coagulopathy prior to his liver and bone marrow biopsies. Within 24 hours after starting FFP, his ferritin level declined by over 50% and his coagulation studies improved.
Although our patient met five out of the eight diagnostic criteria for HLH, because of the dramatic improvement in his serum ferritin after just one dose of FFP, the decision was made to closely monitor him prior to starting stronger immunosuppressive medications.
Patients with inborn metabolism errors [may] present with secondary HLH as an initial manifestation of their underlying metabolic disease.
He did receive a 1 g/kg dose of intravenous immunoglobulin (IVIG) after he recrudesced with fever, but with continued improvement in his serum ferritin. He developed generalized seizures toward the end of his IVIG infusion. It was unclear whether the seizures were a result of HLH, the IVIG itself or a possible inborn metabolism error.
Therefore, he underwent magnetic resonance imaging (MRI) of his brain, which was normal. He was treated with 10 mg/kg of dexamethasone per day, which was tapered as his hyperammonemia, transaminitis and fever improved.
Our patient underwent concurrent biochemical and genetic evaluation for both primary HLH and inborn metabolism errors. An analysis of serum transferrin isoforms was suggestive of a type II congenital disorder of glycosylation (CDG). Rapid whole exome sequencing confirmed his presumed diagnosis by identifying compound heterozygous mutations in the COG4 gene that encodes for part of an oligomeric protein complex involved in Golgi apparatus structure and function.
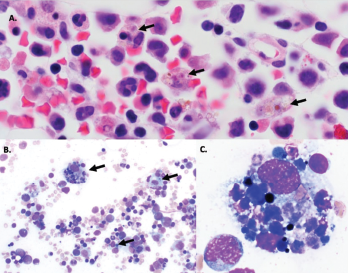
Figure 1A–C: Arrows denote hemophagocytic histiocytes.
Figure A: Hematoxylin and eosin stained bone marrow biopsy (1,000X).
Figure B: May-Gurenwald-Giemsa bone marrow aspirate (100X).
Figure C: May-Gurenwald-Giemsa bone marrow aspirate (1,000X).
COG4 mutations have previously been reported in two patients with autosomal recessive COG4-CDG (CDG-IIj), who were described as having a similar set of clinical symptoms of hypotonia, seizures, coagulopathy and liver dysfunction, as well as recurrent infections.
Over the next three months, the patient experienced two more episodes of fever, liver dysfunction, coagulopathy and shock physiology. The second episode was preceded by a similar set of symptoms of fever, vomiting and diarrhea, and was successfully treated with FFP.
The third episode occurred four days after he received hepatitis A and measles mumps rubella (MMR) vaccinations at 12 months. Our patient again required intensive care and had prolonged hypotension requiring inotropic support. His hypotension resolved after receiving daily FFP, and his hypoxia resolved after a single treatment with protein C concentrate.
Discussion
Congenital disorders of glycosylation are inborn metabolism errors characterized by defects in glycosylation (i.e., the assembly, processing and addition of sugars to proteins and lipids). The two main types of glycosylation are N-linked and O-linked glycosylation.
N-linked glycosylation is the most common covalent protein modification, in which sugars are linked to the nitrogen groups on asparagine residues of proteins in a consensus amino acid sequence (Asn-X-Ser/Thr) with a common pentasaccharide core.1 This modification begins on the cell surface of the endoplasmic reticulum and is completed in the Golgi apparatus.
O-linked glycosylation occurs in the lumen of the endoplasmic reticulum and occurs with the addition of N-acetylgalactosamine (GalNAc) to the hydroxyl group on serine or threonine without the conserved pentasaccharide core.2
Because glycosylation plays an important role in every organ, CDG affects multiple organ systems, including the nervous (causing developmental delays, seizures, ataxia and hypotonia), endocrine, renal and immune systems, as well as the liver (causing transaminitis and coagulopathy).
Looking specifically at the immune system, proper glycosylation is critical for normal immune function. At least 23 CDG patients with immunological involvement, such as an increased propensity to infections and inflammatory and autoimmune complications, have been documented; all of these may demonstrate significant clinical variability associated with the same CDG mutation.3
Manifestations of immune deficiency include recurrent sinopulmonary and gastrointestinal infections, skin abscesses and episodes of bacterial sepsis and invasive fungal infections. Inflammatory and autoimmune manifestations may include an Omenn syndrome-type skin rash, inflammatory bowel disease, celiac disease, autoimmune hypothyroidism, Behçet’s-like disease and familial Mediterranean fever. These manifestations are not mutually exclusive, and patients with immunodeficiency syndromes may also present with autoimmune or inflammatory complications.
With regard to HLH in patients with CDG, case of patients with PMM2-CDG and COG6-CDG presenting with HLH have been reported.4,5 Looking more broadly at patients with inborn metabolism errors, cases of patients with cobalamin C disease, long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHAD) and lysinuric protein intolerance who presented with HLH have also been reported.6-8
Whether these patients presented with HLH as a direct result of their underlying metabolic disease is not certain. However, it has been hypothesized the increased buildup of toxic metabolites related to the underlying metabolic defect may decrease NK cell function and increase cell expansion and activation of macrophages.9
In Sum
Our patient is the third known patient with COG4-CDG (CDG-IIj) and represents a novel presentation of CDG due to COG4 defect with associated immune dysfunction manifesting as HLH and subsequent recurrent episodes of inflammation presenting with shock physiology.
CDG and inborn metabolism errors should be considered during diagnostic evaluation for patients with hemophagocytic lymphohistiocytosis symptoms, especially in very young patients who have previous evidence of developmental delays, neurologic symptoms (e.g., seizures and hypotonia), and liver dysfunction manifesting as coagulopathy, elevated serum transaminases and elevated serum ammonia.
CDG patients may also develop acute episodes of severe inflammation in the absence of cellular regulatory defects, for which FFP and protein C concentrate may have therapeutic value.
 Jeffrey Lo, MD, is a third-year fellow in pediatric rheumatology at Boston Children’s Hospital.
Jeffrey Lo, MD, is a third-year fellow in pediatric rheumatology at Boston Children’s Hospital.
References
- Lyons JJ, Milner JD, Rosenzweig SD. Glycans instructing immunity: The emerging role of altered glycosylation in clinical immunology. Front Pediatr. 2015 Jun 11;3:54.
- Monticelli M, Ferro T, Jaeken J, et al. Immunological aspects of congenital disorders of glycosylation (CDG): A review. J Inherit Metab Dis. 2016 Nov;39(6):765–780.
- Pascoal C, Francisco R, Ferro T, et al. CDG and immune response: From bedside to bench and back. J Inherit Metab Dis. 2020 Jan;43(1):90–124.
- Noelle V, Knuepfer M, Pulzer F, et al. Unusual presentation of congenital disorder of glycosylation type 1a: Congenital persistent thrombocytopenia, hypertrophic cardiomyopathy and hydrops-like aspect due to marked peripheral oedema. Eur J Pediatr. 2005 Apr;164(4)223–226.
- Althonaian N, Alsultan A, Morava E, Alfadhel M. Secondary hemophagocytic syndrome associated with COG6 gene defect: Report and review. JIMD Rep. 2018;42:105–111.
- Wu S, Gonzalez-Gomez I, Coates T, Yano S. Cobalamin C disease presenting with hemophagocytic lymphohistiocytosis. Pediatr Hematol Oncol. 2005 Dec;22(8):717–721.
- Erdol S, Ture M, Baytan B, et al. An unusual case of LCHAD deficiency presenting with a clinical picture of hemophagocytic lymphohistiocytosis: Secondary HLH or coincidence? J Pediatr Hematol Oncol. 2016 Nov;38(8):661–662.
- Barilli A, Rotoli BM, Visigalli R, et al. Impaired phagocytosis in macrophages from patients affected by lysinuric protein intolerance. Mol Genet Metab. 2012 Apr;105(4):585–589.
- Taurisano R, Maiorana A, de Benedetti F, et al. Wolman disease associated with hemophagocytic lymphohistiocytosis: attempts for an explanation. Eur J Pediatr. 2014 Oct 173(10):1391–1394.