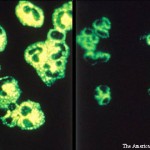
Case
Click to enlarge.
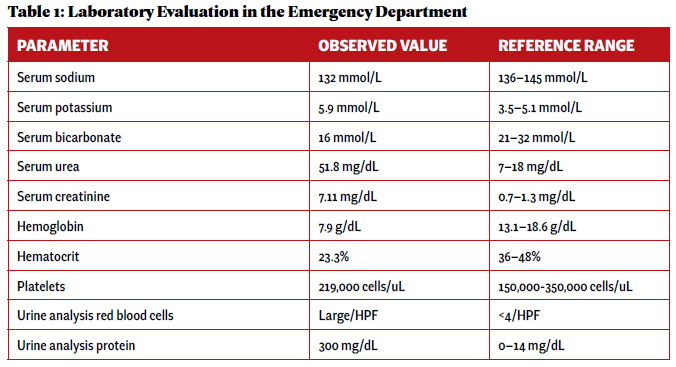
An otherwise healthy 23-year-old woman presented to her primary care provider with gross hematuria initially attributed to her menstrual cycles. Over the next five months, she developed edema, hemoptysis, headaches, nausea and vomiting, and experienced episodes of epistaxis prompting evaluation at an emergency department. Pertinent initial laboratory results on hospital admission are presented in Table 1.
Laboratory evaluation demonstrated acute renal failure, 2+ proteinuria and gross hematuria. In addition, evaluation in the emergency department was significant for multi-focal pulmonary opacities seen on a computed tomography (CT) scan of the chest (see Figure 1).
Her clinical presentation was ultimately consistent with a pulmonary-renal syndrome, with features of diffuse alveolar hemorrhage and renal failure, and she was transferred to our hospital for inpatient consultations with a nephrologist and a rheumatologist.
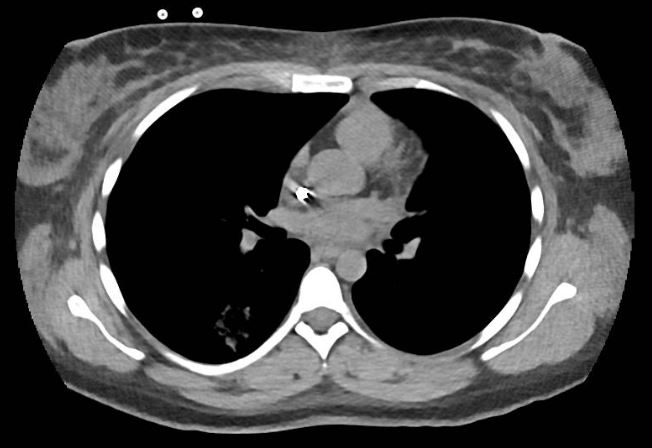
FIGURE 1: Computed tomography of the patient’s chest revealed multi-focal opacities. (Click to enlarge.)
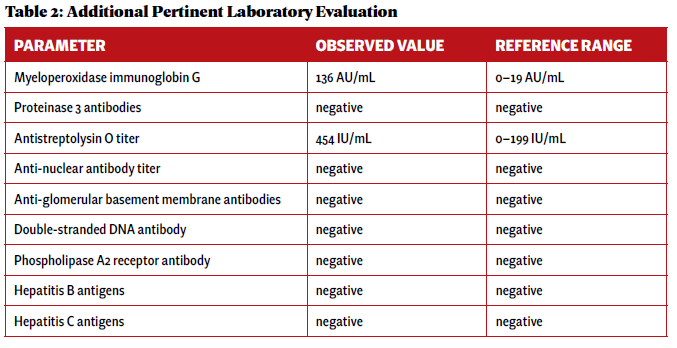
An additional laboratory evaluation was performed at a different hospital to assess the etiology of her pulmonaryrenal syndrome (see Table 2).
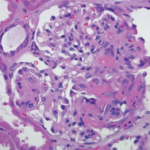
A renal biopsy was performed due to suspected vasculitis. Biopsy results demonstrated a rapidly progressive glomerulonephritis secondary to ANCA-associated vasculitis, with active crescentic and necrotizing glomerulonephritis, 50% sclerosis and some mesangial immunoglobin A (IgA ) deposition. With the presence of multiple crescents in the context of antimyeloperoxidase (anti-MPO) positivity and minimal immunofluorescence deposition, ANCA-associated crescentic glomerulonephritis with superimposed mild IgA nephropathy was favored as a diagnosis.
The patient was ultimately diagnosed with ANCA-associated vasculitis in the setting of MPO positivity and pauci-immune glomerulonephritis, as demonstrated on renal biopsy.
Click to enlarge.
The patient was initiated on pulse-dose corticosteroids, plasmapheresis and supportive dialysis, and 1,000 mg of rituximab separated by 14 days. Seven cycles of plasmapheresis were initially anticipated, but the therapy was stopped after two cycles due to a notable response to the corticosteroid treatment. The patient was discharged home with a corticosteroid taper and continued on supportive dialysis in the outpatient setting.
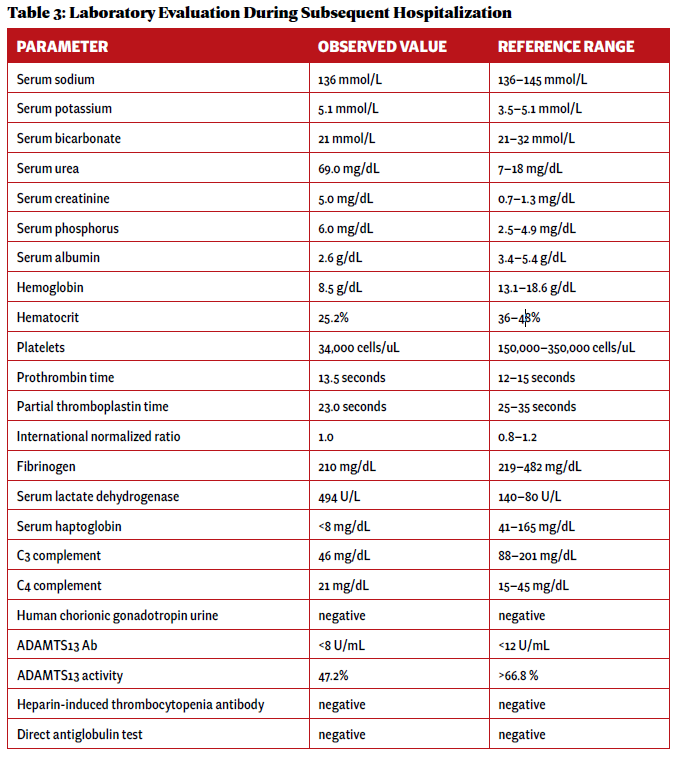
Two weeks after initial hospital discharge, the patient returned to an emergency department complaining of worsening abdominal and bilateral flank pain. Additional imaging was ultimately negative for new thrombosis or infarction, and her symptoms were attributed to volume overload in the setting of dialysis. Laboratory testing incidentally revealed new-onset thrombocytopenia and low-grade schistocytes (see Table 3).
Click to enlarge.
These laboratory findings and her clinical presentation were ultimately consistent with microangiopathy hemolytic anemia and thrombocytopenia, collectively a thrombotic microangiopathy syndrome. In the setting of low complement levels with no additional features of an overlap rheumatic syndrome, a diagnosis of c-TMA was made.
Evaluation was without findings for additional etiologies in the differential diagnosis for a TMA syndrome, including thrombotic thrombocytopenic purpura (TTP); heparin-induced thrombocytopenia; hemolysis, elevated liver enzymes, low platelet count (HELLP) syndrome; eclampsia; drug-induced TMA; scleroderma renal crisis; antiphospholipid antibody syndrome; malignant hypertension; Shiga toxin-producing Escherichia coli infection; and malignancy.
Anti-complement therapy and plasmapheresis were initially considered, but the patient’s platelets continued to increase along with a slow resolution of the c-TMA syndrome without additional intervention. Further escalation of immunosuppressive therapy was, therefore, deferred.
The patient was ultimately discharged five days later and continued on a prolonged, moderate-dose corticosteroid taper. Two weeks later, laboratory evidence indicated complete resolution of the c-TMA syndrome.
Discussion
ANCA-associated vasculitis is a rare autoimmune condition characterized by inflammation of small and medium-sized vessels due to antibodies to either proteinase 3 or MPO. Subtypes of ANCA-associated vasculitis include microscopic polyangiitis (MPA), granulomatous polyangiitis (GPA), and eosinophilic granulomatous polyangiitis (EGPA). Multi-organ dysfunction, including renal and respiratory impairment, is a typical feature of ANCA-associated vasculitis.1
TMA is characterized by microangiopathy hemolytic anemia and thrombocytopenia, which can also demonstrate varying degrees of multi-organ dysfunction resembling ANCA-associated vasculitis. c-TMA, also known as atypical hemolytic uremic syndrome (aHUS), is a subtype of TMA. The pathogenesis of c-TMA is secondary to either a hereditary or acquired dysfunction of proteins, resulting in dysregulation of the alternative pathway of the complement cascade.2 The excessive activation of the complement cascade results in uncontrolled damage to vascular endothelium, causing the hypocomplementemia and ischemic renal injury that are hallmarks of its clinical presentation.3
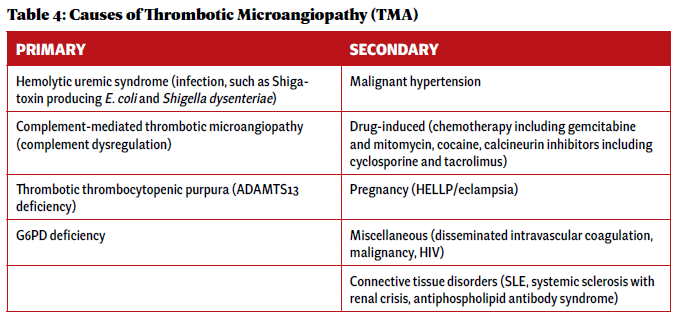
c-TMA is ultimately a clinical diagnosis of exclusion. In the initial evaluation of a suspected TMA syndrome, it is essential to rule out primary TMA syndromes (including TTP and aHUS) and evaluate for systemic disease as a cause of secondary TMA (see Table 4).
Click to enlarge.
The coexistence of ANCA-associated vasculitis with c-TMA is extremely rare. The exact prevalence and incidence are unknown, with only five case reports found in the current medical literature.4 The pathogenesis of ANCA-associated vasculitis is not fully elucidated, and the complement cascade, especially the alternative pathway, has a role in the amplification of continuing neutrophil chemotaxis and priming within the disease process. Moreover, avacopan, a C5a receptor antagonist, has demonstrated some effectiveness at inducing ANCA-associated vasculitis disease remission while limiting the use of glucocorticoids.
Typical treatment of ANCA-associated vasculitis includes glucocorticoids and either rituximab or cyclophosphamide. However, in the case of severe ANCA-associated vasculitis with a coexisting c-TMA syndrome and progressive multi-organ dysfunction, it is essential to consider the use of anti-complement therapy, such as avacopan, eculizumab and ravulizumab.
Terminal complement inhibition via eculizumab or ravulizumab, via inhibition of C5, prevents cleavage of C5 into C5a and C5b, preventing generation of the terminal complement complex C5b-9.
A theoretical advantage of inhibiting C5a, as compared with C5, is that it provides a pathway for C5b-9 to remain functional, potentially conserving some aspect of the innate immune system, especially toward encapsulated bacteria.
In the case of c-TMA, terminal complement C5b-9 would likely best be targeted due to its roles in intravascular hemolysis, as seen in other disease states, such as paroxysmal nocturnal hemoglobinuria. Additionally, the use of anti-complement therapy in c-TMA syndromes has demonstrated a lower risk of progression to end-stage renal disease.5
ANCA-associated vasculitis is characterized by various clinical syndromes, commonly renal and pulmonary complications. TMA syndromes in the ANCA-associated vasculitis setting are considered rare and poorly described. It is essential to evaluate for primary TMA syndromes and other systemic causes.
 Logan Oliver, MD, graduated from McGovern Medical School at UTHealth, Houston, and is currently an internal medicine resident at Naval Medical Center, San Diego.
Logan Oliver, MD, graduated from McGovern Medical School at UTHealth, Houston, and is currently an internal medicine resident at Naval Medical Center, San Diego.
 Jason J. Weiner, MD, FACP, FACR, is a rheumatologist and currently serves as chair of the Department of Medicine at Naval Medical, Center, San Diego. He completed his rheumatology fellowship at Duke University, Durham, N.C., and has research interests in health professions education.
Jason J. Weiner, MD, FACP, FACR, is a rheumatologist and currently serves as chair of the Department of Medicine at Naval Medical, Center, San Diego. He completed his rheumatology fellowship at Duke University, Durham, N.C., and has research interests in health professions education.
References
- Austin K, Janagan S, Wells M, et al. ANCA-associated vasculitis subtypes: Recent insights and future perspectives. J Inflamm Res. 2022 Apr 21;15:2567–2582.
- Fleming P, Cheung M, Sokol D. Complement-mediated thrombotic microangiopathy: A murky presentation of a rare disease entity. Blood. 2018 Nov;132(Supp 1):5005–5005.
- Kościelska-Kasprzak K, Bartoszek D, Myszka M, et al. The complement cascade and renal disease. Arch Immunol Ther Exp (Warsz). 2014 Feb;62(1):47–57.
- Shukla S, Sekar A, Naik S, et al. ANCA-associated vasculitis with systemic thrombotic microangiopathy: A review of literature. Indian J Nephrol. 2024 Mar–Apr;34(2):155–161.
- Huizenga N, Zonozi R, Rosenthal J, et al. Treatment of aggressive antineutrophil cytoplasmic antibody-associated vasculitis with eculizumab. Kidney Int Rep. 2019 Dec 4;5(4):542–545.