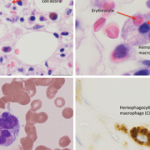
An activated macrophage.
David Scharf/Science Source
Primary hemophagocytic lymphohistiocytosis (HLH) is caused by genetic mutations and inherited syndromes; it therefore occurs in the pediatric age group. Secondary HLH, however, is more common in adults and is often triggered by other disease states, such as malignancies, chronic immunosuppression, infections and autoimmune disease.1,2 Macrophage activation syndrome (MAS) is a subset of secondary HLH seen in patients with autoimmune diseases, such as systemic juvenile idiopathic arthritis (sJIA), systemic lupus erythematosus (SLE) and adult-onset Still’s disease.3
A high index of suspicion for HLH is crucial because treatment delay is associated with high mortality and morbidity. Excluding other causes of the symptoms also prevents inappropriate treatment, which can lead to poor outcomes.
The patient had a 14-year history of anti-neutrophil cytoplasmic antibody (ANCA) associated vasculitis (AAV). She had presented with glomerulonephritis, which was treated with cyclophosphamide and glucocorticoids, followed by azathioprine. She reported that her current complaints were similar to the symptoms she experienced when she was first diagnosed with AAV.
| LAB VALUE | Day 1 | Day 2 |
|---|---|---|
| Sodium (135–145 mmol/L) | 129 mmol/L (L) | 132 mmol/L (L) |
| Carbon dioxide (21–32 mmol/L) | 22 mmol/L | 22 mmol/L |
| BUN (7–18 mg/dL) | 22 mg/dL(H) | 24 mg/dL (H) |
| Creatinine (0.60–1.30 mg/dL) | 1.60 mg/dL(H) | 1.7 mg/dL (H) |
| Lactic acid (0.36–1.7 mmol/L) | 1.64 mmol/L | |
| AST (15–37 U/L) | 522 U/L (H) | 717 U/L (H) |
| ALT (13–61 U/L) | 228 U/L (H) | 292 U/L (H) |
| Alk Phosphatase (45–117 U/L) | 168 U/L (H) | 163 U/L (H) |
| Albumin (3.4–5.0 mg/dL) | 2.8 mg/dL (H) | 2.6 mg/dL (L) |
| WBC (4.5–10.5 10*3/uL) | 3.96 K/uL (L) | 3.92 K/uL( L) |
| Band neuts % (2–8%) | 0.03 | 14% (H) |
| Hgb (11.4–15.5 g/dL) | 11.6 g/dL (L) | 10.9 g/dL (L) |
| Hct (37.0–47.0%) | 32.2% L | 31.0% L |
| Platelet count (130–385 10*3/uL) | 38 K/uL (L) | 36 K/uL (L) |
| ESR (0–20 mm/HR) | 22 mm/HR (H) | |
| CRP (<0.300 mg/dL) | 18.1mg/dL (H) | |
| Total creatinine kinase (21-215 U/L) | 156 U/L | |
| LDH (84–246 U/L) | 1,275 U/L (H) | |
| Monoscreen | Negative | |
| Influenza Type A and B Ag | Negative | |
| Hepatitis panel (Hepatitis A IgM, HBsAg, Hep B Core IgM, Hepatitis C Ab | Negative | |
| HIV 1&2 Antibody | Negative | |
| HIV p24 Antigen | Negative |
Her home medication use included azathioprine 100 mg daily, amlodipine 5 mg daily and 25 mg of metoprolol twice daily.
At the time of presentation, her temperature was 102.6° F, and she had a blood pressure of 83/63 mmHg and a heart rate of 102/minute. Her respiratory rate was 19 breaths/minute with oxygen saturation of 93% on room air.
She was oriented, but appeared uncomfortable. Her mucosal membranes were dry, and she was tachycardiac; her examination was otherwise not revealing.
Her laboratory tests were consistent with shock (see Table 1, left). A computerized tomography (CT) scan of her chest demonstrated small pleural effusions with bibasilar infiltrates, but no lymphadenopathy. An abdominal ultrasound confirmed her liver was grossly unremarkable, with no dilatation of the common bile duct, no gall stones, ascites or splenic enlargement.
Diagnosis & treatment: Her admitting diagnosis was septic shock, and she was treated with intravenous vancomycin and cefepime. An infectious disease specialist was consulted to evaluate the patient for atypical infections. The patient’s blood and urine cultures did not identify an organism, so she was transitioned to azithromycin.
During her admission, she had a persistent, high-grade fever, with a maximum temperature of 103.6° F. Her respiratory status continued to worsen, with tachypnea and hypoxia requiring increasing oxygen supplementation. Her laboratory test demonstrated leukopenia, bandemia, thrombocytopenia, transaminasemia and declining renal function. She was noted to have a discordance between her erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP), and tests for infectious etiologies, including viral hepatitis, human immunodeficiency virus and parvovirus were all negative.
She was treated empirically with 500 mg of intravenous methylprednisolone to address the possibility that she was experiencing a relapse of her underlying rheumatic disease, and she was transferred to our center for rheumatology input.
Upon arrival, she was noted to have a persistent fever and respiratory distress; she was tachypneic and was using accessory muscles despite high-flow oxygen therapy. She was subsequently intubated for respiratory failure.
A repeat chest X-ray showed worsening bilateral pulmonary infiltrates, and she was restarted on vancomycin, cefepime and doxycycline. She was also given pulse-dose methylprednisolone 1 gm daily for three days.
Further laboratory testing (see Table 2, below) again showed discordance between her ESR and CRP, prompting the consideration of alternative diagnoses to sepsis and vasculitis. Bronchoalveolar lavage failed to demonstrate evidence of diffuse alveolar hemorrhage (DAH), infection or malignancy. She subsequently developed altered mental status, which was not felt to be due to infection or non-convulsive status epilepticus.
| LAB COMPONENT | day 3 |
|---|---|
| Sodium (135–145 MMol/L) | 139 MMol/L |
| BUN (6–20 mg/dL) | 31 mg/dL (H ) |
| Creatinine (0.5–1.2 mg/dL) | 1.99 mg/dL (H) |
| AST (10–42 IU/L) | 626 IU/L (H) |
| ALT (10–60 IU/L) | 245 IU/L (H) |
| ALP (42–121 IU/L) | 175 IU/L (H) |
| WBC (4.0–10.5 K/uL) | 4.6 K/uL |
| Hemoglobin (12.0–16.0 g/dL ) | 11.5 g/dL (L) |
| Hematocrit (36–46%) | 31.7% (L) |
| Platelets (130–400 K/uL) | 50,000 K/uL (L) |
| ESR (0–30 mm/HR) | 5 mm/HR |
| CRP (<1.0 mg/dL) | 14.9 mg/dL (H) |
| LDH (135–214 IU/L) | 2010 IU/L (H) |
| Ferritin (13–150 ng/mL) | 54,059 ng/mL (H) |
| Triglycerides (<150 mg/dL) | 638 mg/dL (H) |
| Fibrinogen (204–475 mg/dL) | 199 mg/dL (L) |
| ANCA screen (C-ANCA and P-ANCA titer) | Negative |
| Rheumatoid factor | <14 IU/mL |
| Ribosomal protein | <1.0 NEG AI |
| Anti-DNA, native double strand | <1 IU/mL |
| Sm/RNP | <1.0 NEG AI |
| Scleroderma (SCL-70) | <1.0 NEG AI |
| Sjögren’s antibody SSA | <1.0 NEG AI |
| Sjögren’s antibody SSB | <1.0 NEG AI |
| Anti-Smith antibody | <1.0 NEG AI |
| Anti-centromere B antibody | <1.0 NEG AI |
| Anti Jo-1 antibody | <1.0 NEG AI |
| Anti-Streptolysin O | <50 Ref: <200 IU/mL |
| Anti-nuclear antibody | Negative |
| CCP Ab IgG | 77 H, Ref: strong positive >59 |
| Complement C3 | 70 L, Ref: 90–180 mg/dL |
| Complement C4 | 32.8, Ref: 10–40 mg/dL |
| Thyroid peroxidase (TPO) Ab | <1, Ref: <9 IU/mL |
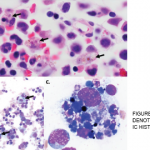
A bone marrow biopsy was obtained and demonstrated a clonal T cell population, with megakaryocytic hyperplasia and rare hemophagocytosis (see Figure 1, opposite). Serum soluble interleukin (IL) 2 receptor (IL-2R) levels were elevated at 10,219 pg/mL (reference range 532–1,891 pg/mL).
A diagnosis of HLH was made, and the antibiotics were stopped. The patient was subsequently treated with 112.5 mg/m2 etoposide (corrected based on renal function) twice weekly for two weeks and 20 mg of intravenous dexamethasone daily for two weeks, after which she gradually improved. She was subsequently extubated, with improvement in mental status, although she still has some memory issues.
She was eventually discharged to a subacute facility for inpatient rehabilitation, where she received 112.5 mg/m2 etoposide weekly for three weeks and 150 mg/m2 of etoposide (corrected based on renal function) weekly for three weeks. She also received dexamethasone, which was tapered from 10 mg daily for two weeks to 5 mg daily for two weeks to 2.5 mg daily for two weeks to 1.25 mg daily for one week, before being discontinued.
Discussion
HLH is broadly divided into two categories: primary HLH, which typically occurs in infants and children, and secondary HLH, which occurs in adults.1 HLH is characterized by an uncontrolled immune response, leading to tissue infiltration by natural killer (NK) cells, cytotoxic T cells and macrophages, and by the release of proinflammatory cytokines, including tumor necrosis factor alpha (TNFα), IL-1 and IL-6, which lead to tissue damage.1
Activated macrophages accumulate in the tissues, including bone marrow, lymph nodes, liver and spleen, which leads to phagocytosis of normal cells—the pathognomonic feature of HLH seen in tissue biopsy.
Primary HLH, including familial HLH (autosomal recessive mutations) and lymphoproliferative syndromes, is caused by genetic mutations impairing the function of NK and T cells. Secondary HLH is usually triggered by an underlying condition, and it may have been related to AAV in our patient.2
Predisposing conditions that have been reported include viral infections (most commonly Epstein-Barr virus), malignancies (leukemias and lymphomas) and rheumatologic illness.
Specific diagnostic criteria for adult-onset HLH are not well established. The diagnosis in adults is based on application of the 2004 pediatric diagnostic criteria (see Table 3, below).4,5 Other clinical and laboratory findings that are supportive of HLH diagnosis in adult population (but are not part of the pediatric diagnostic criteria) include hepatomegaly, transaminitis, elevated LDH, elevated D-dimer with normal INR and PTT, coagulopathy, elevated CRP, hyponatremia, rash and neurologic involvement.2 Genetic mutations are part of the diagnostic criteria, but are not routinely tested in adults.
| DIAGNOSTIC CRITERIA FOR HLH | ||
|---|---|---|
| Molecular tests | ||
| Genetic mutation | PRF1 | |
| UNC13D | ||
| TX11 | ||
| STXBP2 | ||
| RAB27 | ||
| XLP | ||
| Or | ||
| 5 of 8 clinical criteria | Fever | Peak temperature of >38.5°C |
| for >7 days | ||
| Splenomegaly | Spleen palpable >3 cm below costal margin | |
| Cytopenias of at least two lineages in the peripheral blood | Hemoglobin <9 g/100 mL | |
| Platelets <100 X 103/mL | ||
| Neutrophils <1 X 103/mL | ||
| Hypertriglyceridemia | Fasting, ≥265 mg/100 mL | |
| and/or hypofibrinogenemia (≤150 mg/100 mL) | ||
| Hemophagocytosis in bone marrow (BM), spleen or lymph nodes | Low or absent NK cell activity | |
| Ferritin ≥500 ng/mL | ||
| Low or absent natural killer cell activity | ||
| Soluble CD25 (that is, soluble IL-2 receptor) >2,400 U/mL | ||
| PRF1: Perforin protein; UNC13D: Munc 13-4 protein; STX11: Syntaxin 11 protein; STXBP2: Munc 18-2 protein. |
Early in the disease process, HLH can be overlooked, especially given the nonspecific nature of the symptoms and its association with other conditions. Clinical findings of HLH are similar to those of systemic inflammatory response syndrome (SIRS) and are often initially treated as an infection, as in this case. Hyperferritenemia with values >10,000 ng/mL is 90% sensitive and specific in the pediatric population. In adults, however, hyperferritenemia is less specific. In fact, levels as high as 50,000 ng/mL have been reported in infections, hepatocellular injury, renal failure and other conditions.6
Relative changes in overall clinical and laboratory parameters is a good way to make an early diagnosis of MAS.9 In our patient’s case, there was a drop in ESR and a discordance between ESR and CRP levels. This discordance is often found in early cases of MAS.9
Although pathognomonic, hemophagocytosis is neither sensitive nor specific for HLH.1 Hemophagocytosis may not be seen in a bone marrow aspiration or biopsy in early stages of HLH. Sometimes, even when not seen on bone marrow examination, autopsy examination of the spleen or lymph nodes may reveal hemophagocytosis. Bone marrow biopsy should be performed when considering a diagnosis of HLH, not only for diagnosis but also to exclude hematological malignancy as the trigger.1,6,7 Repeating it may also be necessary in later disease stages.
Soluble IL-2 receptor CD 25 has recently been reported as a low-cost diagnostic test for HLH in adults. Along with ferritin and lactate dehydrogenase (LDH), the IL-2 receptor assay is a useful way to assess treatment response. However, IL-2 receptor testing is mostly performed in reference laboratories, and it may take several days to obtain results.1,2
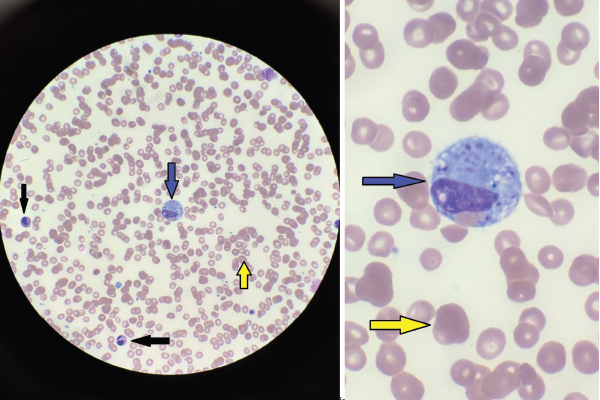
Figures 1A & 1B
Figures A (left; 40x magnification) and B right; (100x magnification). The bone marrow aspiration smear shows activated macrophages with phagocytosis of hematopoietic cells (blue arrow), including mature red blood cells (yellow arrows) and neutrophils (black arrows).
HLH probability score (H score), a freely available online calculator, was based on a multicenter, retrospective cohort of adult patients and can be used to calculate the diagnostic likelihood of HLH in patients with a constellation of clinical and laboratory features suggestive of HLH.8
The treatment of HLH in adults is derived from HLH-94 protocols developed for children and includes corticosteroids, etoposide, cyclosporin A and intrathecal therapy for patients with central nervous system (CNS) involvement.11 For patients with MAS, in contrast to other patients with secondary HLH, pulse-dose methylprednisolone (1 g/day) is the first-line treatment.
For patients who do not respond to high-dose steroids, the addition of cyclosporine at a dose of 2–7 mg/kg/day is recommended. For patients refractory to both steroids and cyclosporine, treatment protocols including etoposide should be considered.9 In our case, etoposide was started under the direction of the hematology team and the patient had a good response.
Alternative treatments for patients with MAS include intravenous immunoglobulin (IVIG), plasma exchange, cyclophosphamide and biologic agents, such as IL-1 inhibitors.9,11 Hematopoietic cell transplant (HCT) should be considered for patients with familial orgenetic mutations for HLH, underlying hematologic malignancies and CNS involvement.11
Secondary infection due to iatrogenic depletion of leukocytes is a major cause of mortality in adults with HLH. Patients being treated with HLH-specific therapies should receive intensive nursing care, neutropenic precautions and prophylaxis against opportunistic infections.11
Primary HLH, including familial HLH (autosomal recessive mutations) & lymphoproliferative syndromes, is caused by genetic mutations impairing the function of NK & T cells. Secondary HLH is usually triggered by an underlying condition.
Conclusion
HLH is rare in adults and associated with very high mortality rates, especially if associated with malignancy. A high index of suspicion in patients with symptoms of unexplained SIRS, multiple organ dysfunction syndrome and laboratory abnormalities is essential to reduce mortality. A very important indicator is a discordance between ESR and CRP and, as in our case, should raise suspicion of HLH. Prompt treatment can result in good outcomes for patients with this life-threatening disease—as in our case.
 Srujana Pachigolla, MD, is a board-certified internist and hospitalist for Carilion Roanoke Memorial Hospital, Va.
Srujana Pachigolla, MD, is a board-certified internist and hospitalist for Carilion Roanoke Memorial Hospital, Va.
 Adegbenga Bankole, MD, is chief of rheumatology at the Carilion Clinic and is an associate professor of medicine at Virginia Tech Carilion School of Medicine, Roanoke, Va.
Adegbenga Bankole, MD, is chief of rheumatology at the Carilion Clinic and is an associate professor of medicine at Virginia Tech Carilion School of Medicine, Roanoke, Va.
References
- Madkaikar M, Shabrish S, Mukesh Desai M. Current updates on classification, diagnosis and treatment of hemophagocytic lymphohistiocytosis (HLH). Indian J Pediatr. 2016 May;83(5):434–443.
- Hayden A, Park S, Giustini D, et al. Hemophagocytic syndromes (HPSs) including hemophagocytic lymphohistiocytosis (HLH) in adults: A systematic scoping review. Blood Rev. 2016 Nov;30(6):411–420.
- Kumar V, Sharma B, Nigam AS. Case series of hemophagocytic lymphohistiocytosis from a tertiary care centre: An underdiagnosed entity. Turk Patoloji Derg. 2019 Apr 12; 35(3):207–212.
- Henter JI, Elinder G, Söder O, Ost A. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr Scand. 1991 Apr;80(4):428–435.
- Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007 Feb;48(2):124–131.
- Meena NK, Sinokrot O, Duggal A, et al. The performance of diagnostic criteria for hemophagocytic lymphohistiocytosis in critically ill patients. J Intensive Care Med. 2019 Mar 12:885066619837139.
- Gupta A, Weitzman S, Abdelhaleem M. The role of hemophagocytosis in bone marrow aspirates in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008 Feb;50(2):192–194.
- Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014 Sep;66(9):2613–2620.
- Lerkvaleekul B, Vilaiyuk S. Macrophage activation syndrome: early diagnosis is key. Open Access Rheumatol. 2018 Aug 31;10:117–128.
- Bracaglia C, Prencipe G, De Benedetti F. Macrophage activation syndrome: Different mechanisms leading to a one clinical syndrome. Pediatr Rheumatol Online J. 2017 Jan 17;15(1):5.
- La Rosée P, Horne A, Hines M, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019 Jun 6;133(23):2465–2477.