Although the destructive changes consistent with rheumatoid arthritis (RA) are said to have been identified in the skeletal remains of North American natives from more than 4,000 years ago, RA was first described in Europe around 1800, just after the start of the Industrial Revolution. Potentially relevant to the pathogenesis of RA, the Industrial Revolution resulted in the pollution of air, water, and soil. It is only recently that studies have started to identify how genes and the environment may interact, resulting in RA. Studies are beginning to shed light on the mechanisms by which structural and regulatory components of the joint may directly contribute to the progression of the disease by serving as fuel for the flames of the inflammatory process mediated through Toll-like receptors (TLRs).
Toll-like Receptors
In the early 1980s, the effects of chemical mutations in Drosophila melanogaster resulted in alterations in the development of the resulting embryos, one of which was referred to as “toll,” meaning fantastic, amazing, or cool, by Nobel laureate Christine Nusslein-Volhard.1 Soon afterwards, Carl Hashimoto and colleagues identified and characterized the gene responsible for this mutation, called the Toll gene. Toll was identified as a protein with a transmembrane domain and an ectodomain containing multiple leucine repeat segments. Therefore, Toll apparently interacts with binding partners within the embryo to direct normal development. I will come back to this point when discussing the pathogenesis of RA. Subsequent studies by Jules Hoffman, who received a Nobel Prize in 2011, also demonstrated that Drosophila with Toll deleted were at increased risk to develop fungal infections, suggesting that Toll is also capable of interacting with microbial ligands.
In 1994, the first human gene that resembled Toll was identified by Nobuo Nomura and colleagues, and in 1997, Ruslan Medshitov and Charles Janeway identified that the human Toll receptor was capable of activating NF-kB and inducing proinflammatory cytokines, which we know are important in the pathogenesis of RA. The product of the human Toll gene has subsequently come to be known as the TLR. In 1998, the group, led by Bruce Beutler, who also was awarded a Nobel Prize in 2011, identified that lipopolysaccharide (LPS) from gram-negative, bacteria-activated TLR4. Subsequently, a total of 10 human TLRs have been characterized.
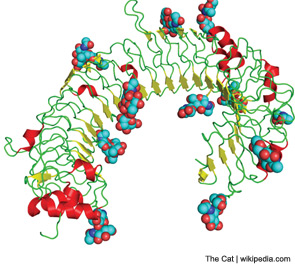
The function of TLRs can be separated based on the location of their expression within the cell either extracellularly or within the endosome (see Figure 1). Although TLRs are critical for the function of monocytes, macrophages, dendritic cells, and natural killer cells, they are also expressed by other cell types, such as fibroblasts and endothelial cells. Ligation of TLRs results in the activation of NF-kB and mitogen-activated protein kinases including Jun N-terminal Kinase, p38, and extracellular signal-regulated kinase, which promote the expression of proinflammatory cytokines, chemokines, and matrix metalloproteinases. TLRs expressed on antigen presenting cells such as macrophages and dendritic cells contribute to the induction of adaptive immunity by T and B lymphocytes by contributing to the maturation of dendritic cells to promote efficient antigen presentation and by the release of cytokines such as interferons, interleukin (IL) 1β, and IL-6, which promote lymphocyte differentiation and activation. The induction of autoantibodies in RA, including rheumatoid factor and antibodies to citrullinated proteins (ACPAs) have been shown to require antigen-presenting cells, suggesting that TLRs are involved in the initiation of autoimmunity in RA.2 How TLRs might contribute to the persistence or progression of RA will be discussed below.
Genetic Background
The genetic background associated with RA first came to light in 1976, when Peter Stastny performed allogeneic mixed lymphocyte cultures and found that the cells from patients with RA were less likely to activate cells from other patients with RA than were cells from unrelated individuals without RA.3 This suggested that there were similarities in the major histocompatibility complex (MHC) region that were shared by patients with RA. These studies led to the identification that HLA-DRB1 alleles, which include shared epitopes (SEs), are associated with RA.4 The SEs are enriched in positively charged arginines around the antigen-binding grove of the third hypervariable region of a subset of HLA-DRB1 alleles that included *0401, *04014, and *0101. A SE is present in about 60% to 70% of patients with RA. More recent studies have identified that the SE is more strongly associated with ACPAs than with the presence of RA itself.5 A mechanism contributing to the production of ACPAs by SE-positive individuals appears to be related to the increased avidity of binding of citrullinated peptides to the SE. Citrullination is a posttranslational modification mediated by peptidyl arginine deiminase, which converts the positively charged amino group on arginine to a neutral oxygen. This increased binding appears to result in the enhanced ability of citrullinated peptides to bind to SEs on antigen-presenting cells, resulting in the activation of T and B lymphocytes and in the induction of ACPAs. However, the presence of SE alleles is not uncommon, detected in almost 30% of the Caucasian population of European descent and 60% of the patients with RA.6