Epidemiology
KD is a disease of young children, with the majority of cases occurring in children younger than 5 years old. Although it has been described throughout the world, a genetic susceptibility likely underpins the higher incidence in Japanese and Korean children compared with children in the U.S. and European countries.
Epidemiologic data has been collected in Japan every two years since 1970, with the most recent KD incidence rate documented at 239.6 per 100,000 children age 0–4 years in 2010. The incidence rate of KD among South Korean children in 2011 was 134.4 per 100,000 in children younger than 5 years old. In contrast, the U.S. hospitalization rate for KD in children younger than 5 years old was 20.8 per 100,000 in 2006.
It should be noted that although KD is less common in very young babies under 6 months old and children who are older than 5 years old, these children are in fact at higher risk for coronary artery involvement, and the diagnosis should be entertained in persistently febrile children in these age groups.
To capture infants at a stage when the disease is treatable, the American Heart Association (AHA) has recommended that children 6 months or younger with fever of seven days or longer duration of unclear etiology who have evidence of systemic inflammation on laboratory studies should undergo echocardiography, regardless of the absence of other stigmata of KD.2
Boys are more likely than girls to be affected by KD and are more likely to have coronary artery involvement. Although it is very unusual for a pair of siblings to have KD concurrently, parents of children with KD in Japan are twice as likely to have had an episode of KD themselves when they were children.
Studies have identified certain demographic features of children at high risk for resistance to treatment with immune intravenous globulin (IVIG) with the concomitant risk of coronary artery aneurysms. Risk factors include male sex, age younger than 6 months and older than 9 years, Asian/Pacific Islander race and Hispanic ethnicity, as well as laboratory findings at the time of diagnosis, including leukocytosis, thrombocytopenia, hypoalbuminemia, hyponatremia, transaminitis and an elevated C-reactive protein.
Risk scores to identify children at risk for IVIG resistance have been constructed and tested in Japanese populations by Kobayashi et al,3 Sano et al4 and Egami et al.5 Unfortunately, these risk scores do not appear to successfully identify North American children at risk for IVIG resistance.6
Etiology
As mentioned above, the etiology of KD remains unknown. The hallmark symptoms of fever and rash mimic an infectious disease, and discrete seasonal peaks with clustering of cases in geographic areas suggest a transmittable vector.7
Further, the epidemiology of KD is consistent with an infectious agent, because it is highly unusual for two populations to have KD: very young babies who may be protected by maternal antibodies via placental transfer, and adults who have presumably seen multiple infectious agents and are immune. However, an infectious agent has been searched for exhaustively. To date no single culprit has been found.
Other theories of the pathogenesis of KD include the possibility of superantigen activity, because children can present with hypotension if myocarditis is significant and there is erythroderma present. However, studies of toxins made by Staphylococcus aureus and Streptococcus pyogenes acting as superantigens have varied in regard to conclusive evidence of such.
Genome-wide association studies have identified polymorphisms in the ITPKC gene (a T cell regulator),8 in B lymphoid tyrosine kinase9 and in a high-affinity receptor for immunoglobulin G (FCGR2A) in patients with KD.10 The functional single-nucleotide polymorphisms in the ITPKC gene have been associated with IVIG resistance and worse coronary artery outcomes.
An alternative hypothesis for the pathogenesis of KD is that multiple environmental and/or infectious triggers may lead to a stereotyped inflammatory cascade with the characteristic features of KD as a final common pathway.
Clinical Features & Diagnosis
The diagnosis of KD relies on a case definition (Table 1), because reliable diagnostic testing has not yet been discovered. It should be noted that not all features of KD may be present simultaneously, and a thorough history of the course of the illness is required. All features of KD will resolve, typically within 11–12 days, even in the absence of treatment.
Fever is the cardinal feature of KD and tends to be of abrupt onset. The fevers are typically high, above 39°C (102.2°F), and don’t necessarily remit with antipyretics. According to the classic case definition, fever should be present for longer than four days, although the presence of at least four stigmata of KD on Day 4 of fever permits the diagnosis of KD. Those children who have fewer than four criteria but evidence of coronary artery involvement on echocardiography also meet the case definition. In Japan, children who have four or fewer days of fever but whose illness has been shortened by the administration of IVIG also meet diagnostic criteria.
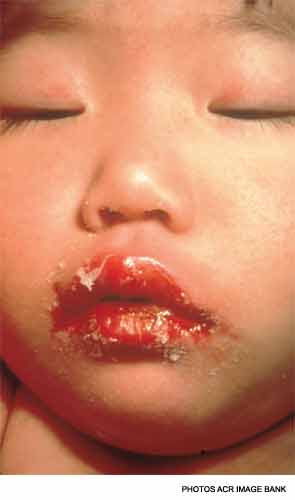
There is no specific rash for KD, although it is commonly a diffuse, maculopapular rash that begins on the trunk. Desquamation in the perineal area is also common in the first week of illness. Morbilliform rashes, erythroderma and erythema multiforme have all been described in patients with KD. Bullous or vesicular rashes are not typical. Conjunctival injection without discharge that spares the limbus (the transitional zone between the cornea and the conjunctiva) is seen in greater than 90% of children with KD.
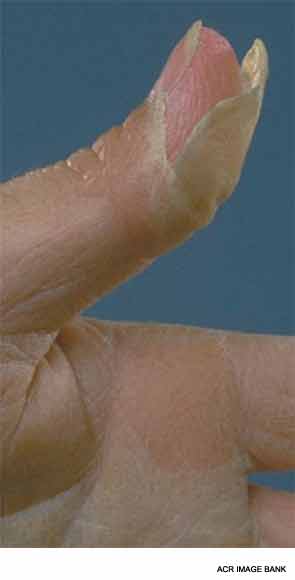
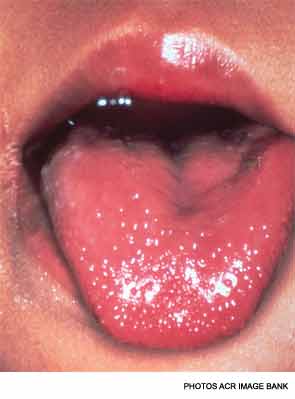
There are several findings in the oropharynx, including a strawberry tongue, a diffusely red oropharynx without ulcers or exudate, and erythematous, cracked lips.
There are two phases to the extremity changes seen in KD: With fever, children have firm swelling of the hands and feet with erythema of the palms and soles. Two to three weeks following fever onset and in the subacute period, periungual peeling, first in the hands and then the feet, appears.
The least commonly seen of the criteria is unilateral cervical chain lymph node swelling, less than 1.5 cm in diameter, which usually appears as a group of matted nodes on imaging.
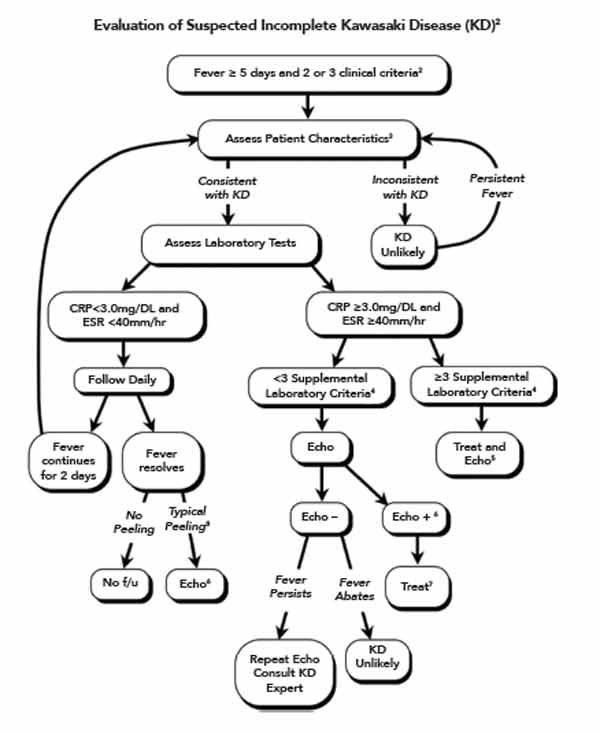
The classic case criteria were developed as a means to perform surveillance of the disease in Japan, and to distinguish KD from acute rheumatic fever and Stevens-Johnson syndrome. It has become clear to clinicians that not all children with KD meet the classic clinical criteria, but some are at higher risk for coronary artery abnormalities (CAA).11 An algorithm utilizing laboratory and echocardiographic data has been developed by the AHA to assist clinicians in diagnosing children with fever of five days or longer and two or three features of KD (Figure 1).2 A retrospective study of patients with KD and coronary artery aneurysms showed that the algorithm correctly identified 97% of children with fewer than four stigmata for treatment with IVIG.12
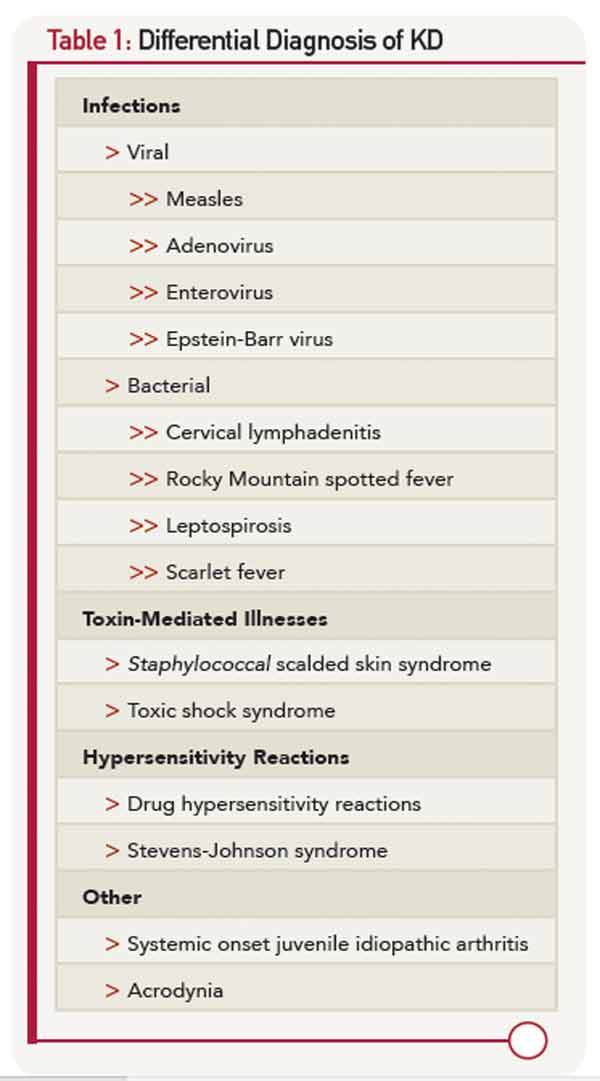
The differential diagnosis of KD (Table 1) is dominated by infectious illnesses commonly seen in childhood. Systemic onset juvenile idiopathic arthritis that is characterized by fever, rash and elevated inflammatory markers can be confused with KD, and in fact, coronary artery dimensions may be enlarged in some of these children. The toxic shock syndromes associated with Streptococcus and Staphylococcus may also mimic KD, although the multisystem involvement can provide clues to a diagnosis other than KD. Stevens-Johnson syndrome and drug hypersensitivity reaction are also on the differential diagnosis, as is the least common mimic, mercury poisoning.
Cardiac Involvement in KD
Echocardiography is the imaging modality of choice in KD, because the coronary arteries in children can be easily visualized with this noninvasive method. Measurements from the inner edge to inner edge of the left main coronary artery (LMCA), left anterior descending artery (LAD), right coronary artery (RCA), left circumflex and posterior descending coronary artery should be assessed. The Japanese Ministry of Health has defined abnormal coronary arteries as greater than 3 mm if the child is younger than 5 years old, and greater than 4 mm if the child is 5 years or older, if the internal diameter is 1.5 times larger than its adjacent segment, or if the lumen is clearly irregular. Because coronary artery dimensions are predicated on the size of the child, coronary artery size adjusted for body surface area, or z scores, are available for the LMCA, proximal LAD and proximal RCA. Because z scores are not available for all coronary artery segments, the Japanese Ministry of Health guidelines are useful in assessing the remainder of the coronary arteries. Lack of tapering of the coronary arteries, left ventricular dysfunction, valvular regurgitation, pericardial effusion and z scores greater than 2.0 may be considered supportive of the diagnosis.
Echocardiograms should be obtained at the time of diagnosis, at approximately two weeks and at six weeks following disease onset if there has been no evidence of coronary artery involvement. Children with coronary artery abnormalities may require more frequent monitoring and should be managed by a pediatric cardiologist. Other imaging modalities, such as CT angiograms, MR angiography or conventional angiography should be decided on a case-by-case basis.
Treatment
Standard treatment for KD includes IVIG and aspirin (ASA). IVIG is given at high doses (2 grams/kg) and should be administered slowly over 8–12 hours to avoid hemodynamic instability. The mechanism by which IVIG effectively treats KD is unclear, but it reduces the risk of coronary artery abnormalities from 20–25% to less than 5%.2 The role of ASA in treating KD is controversial, because studies have shown that it does not influence coronary artery outcomes.13 There is also variation in practice regarding the dosing and duration of ASA. High, anti-inflammatory doses of ASA (80–100 mg/kg/day divided four times per day) are given in North America at least until the child is afebrile for 48 hours followed by low, antiplatelet dosing (3–5 mg/kg/day) for a minimum of six weeks. However, other centers administer high-dose ASA until Day 14 of illness, before switching to low-dose ASA.
Up to 85% of children with KD have full resolution of fever with a single dose of IVIG and do not require further therapy. Those children who develop fevers of 38°C or higher between 36 hours and seven days after completing the first IVIG treatment are said to have IVIG resistance and are at higher risk for CAA.
There are two approaches to treating high-risk children: 1) intensification of primary therapy with corticosteroids or such agents as TNF inhibitors, or 2) providing rescue therapy for those children with persistent or recrudescent fever after initial treatment.
Intensification of primary therapy by the addition of corticosteroids has been tested in the U.S. and in Japan. A randomized trial performed by the Pediatric Heart Network revealed no difference in echocardiographic outcomes in children with KD who received a single pulsed dose of methylprednisolone (30 mg/kg) prior to standard therapy when compared with those who received standard therapy.14 However, a beneficial role for corticosteroids in KD has recently been supported by a well-designed study from Japan called the RAISE study (Randomized Controlled Trial to Assess Immunoglobulin Plus Steroid Efficacy for Kawasaki Disease).15 High-risk children, as defined by a Kobayashi risk score of 5 or higher, were randomized to IVIG or IVIG+ prednisolone arms, with the prednisolone given intravenously for five days at 2 mg/kg/day, followed by an oral taper. The incidence of coronary artery abnormalities was significantly lower in the prednisolone treated group. Additionally, the prednisolone-treated group had fewer days of fever and were less likely to need additional treatments. The differences in outcomes between these two studies may be in part due to the ability of the Kobayashi score to correctly identify children at high risk for CAA and perhaps to the longer duration of corticosteroids.
Intensification of primary therapy with TNF inhibitors has recently been evaluated in a randomized double-blind placebo controlled trial, using a single dose of infliximab (5 mg/kg) + IVIG vs. placebo + IVIG. The results were presented at this year’s ACR/ARHP Annual Meeting by Jane Burns, MD, professor of pediatric infectious diseases at the University of Washington in Seattle. The primary endpoint of differences in rates of IVIG resistance between the two arms was not met. However, there were no differences in adverse events between the two groups. Finally, the group treated with infliximab had more rapid improvement of their inflammatory markers and less fever, but did not have significant differences in their coronary artery outcomes.
In regard to rescue therapy for those patients with IVIG resistance, typically a second dose of IVIG is administered. There is significant variation across centers about the use of other agents, including corticosteroids and infliximab for rescue therapy. Formalized trials will be required to delineate optimal regimens for children with IVIG resistance.
Prognosis of KD
The vast majority of children with KD respond to a single dose of IVIG with rapid clinical improvement and reassuring echocardiography; these children have excellent prognoses. Published mortality rates are quite low, generally less than 1%, and deaths are typically due to rupture of inflamed, dilated coronary arteries within a month of disease onset or myocardial infarction from thrombosed coronary arteries occurring anywhere from a few months to years after disease onset.
The AHA has published specific guidelines for managing children with KD according to their relative risk of myocardial ischemia.2 Children who develop aneurysms require careful long-term monitoring by a pediatric cardiologist. Morbidity can be high in patients with significant cardiac sequelae from KD.
Long-Term Vascular Health
Important yet incompletely answered questions remain regarding those KD patients who never developed evidence of significant coronary artery involvement. Results have been conflicting regarding whether such patients have normal endothelial function or if they are at risk for accelerated atherosclerosis. A recent study offered some reassurance that children with normal coronary artery dimensions or mild ectasia throughout their disease course have similar indices of vascular health as healthy controls.16 However, questions of long-term vascular health in children with KD and normal coronary dimensions will be answered with large, prospective studies of middle-age individuals with a history of KD. Until such data become available, all children with a history of KD should be counseled regarding a heart healthy diet and lifestyle with avoidance of modifiable risk factors for heart disease.
Mary Beth Son, MD, is a specialist in rheumatology at Boston Children’s Hospital.
References
- Kawasaki T. [Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children]. Arerugi = [Allergy]. 1967;16(3):178–222.
- Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: A statement for health professionals from the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation. 2004;110(17):2747–2771.
- Kobayashi T, Inoue Y, Takeuchi K, et al. Prediction of intravenous immunoglobulin unresponsiveness in patients with Kawasaki disease. Circulation. 2006;113(22):2606–2612.
- Sano T, Kurotobi S, Matsuzaki K, et al. Prediction of non-responsiveness to standard high-dose gamma-globulin therapy in patients with acute Kawasaki disease before starting initial treatment. Eur J Pediatr. 2007;166(2):131–137.
- Egami K, Muta H, Ishii M, et al. Prediction of resistance to intravenous immunoglobulin treatment in patients with Kawasaki disease. J Pediatr. 2006;149(2):237–240.
- Sleeper LA, Minich LL, McCrindle BM, et al. Evaluation of Kawasaki disease risk-scoring systems for intravenous immunoglobulin resistance. J Pediatr. 2011;158(5):831–835 e833.
- Yanagawa H, Nakamura Y, Yashiro M, et al. Incidence of Kawasaki disease in Japan: The nationwide surveys of 1999–2002. Pediatr Int. 2006;48(4):356–361.
- Onouchi Y, Gunji T, Burns JC, et al. ITPKC functional polymorphism associated with Kawasaki disease susceptibility and formation of coronary artery aneurysms. Nat Genet. 2008;40(1):35–42.
- Chang CJ, Kuo HC, Chang JS, et al. Replication and meta-analysis of GWAS identified susceptibility loci in Kawasaki disease confirm the importance of B lymphoid tyrosine kinase (BLK) in disease susceptibility. PLoS One. 2013;8(8):e72037.
- Khor CC, Davila S, Breunis WB, et al. Genome-wide association study identifies FCGR2A as a susceptibility locus for Kawasaki disease. Nat Genet. 2011;43(12):1241–1246.
- Ha KS, Jang G, Lee J, et al. Incomplete clinical manifestation as a risk factor for coronary artery abnormalities in Kawasaki disease: A meta-analysis. Eur J Pediatr. 2013;172(3):343–349.
- Yellen ES, Gauvreau K, Takahashi M, et al. Performance of 2004 American Heart Association recommendations for treatment of Kawasaki disease. Pediatrics. 2010;125(2):e234–241.
- Hsieh KS, Weng KP, Lin CC, et al. Treatment of acute Kawasaki disease: Aspirin’s role in the febrile stage revisited. Pediatrics. 2004;114(6):e689–693.
- Newburger JW, Sleeper LA, McCrindle BW, et al. Randomized trial of pulsed corticosteroid therapy for primary treatment of Kawasaki disease. The N Engl J Med. 2007;356(7):663–675.
- Kobayashi T, Saji T, Otani T, et al. Efficacy of immunoglobulin plus prednisolone for prevention of coronary artery abnormalities in severe Kawasaki disease (RAISE study): A randomised, open-label, blinded-endpoints trial. Lancet. 2012;379(9826):1613–1620.
- Selamet Tierney ES, Gal D, Gauvreau K, et al. Vascular health in Kawasaki disease. J Am Coll Cardiol. 2013;62(12):1114–1121.