Diagnosis
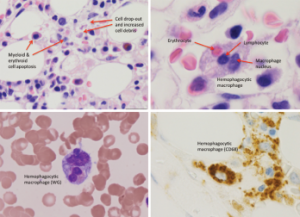
Figure 1: Bone Marrow Biopsy
Hemophagocytosis, with cell apoptosis, cell drop-out and increased cell debris, is shown.
When we met our patient, systemic rheumatic diseases—such as rheumatoid arthritis, systemic lupus erythematosus, systemic sclerosis and mixed connective tissue disease—were considered unlikely given the lack of typical clinical features and negative autoantibody profile. Malignancy also seemed improbable given her negative workup and lack of lymphadenopathy.
Although her CBC showed pancytopenia, the peripheral blood smear was unremarkable. After further discussion with our hematology consultants, a repeat bone marrow biopsy was performed. It demonstrated hypercellularity with trilineage maturing hematopoiesis, stromal damage, cell dropout and increased macrophage phagocytic activity (see Figure 1), which raised the possibility of hemophagocytic lymphohistiocytosis (HLH).
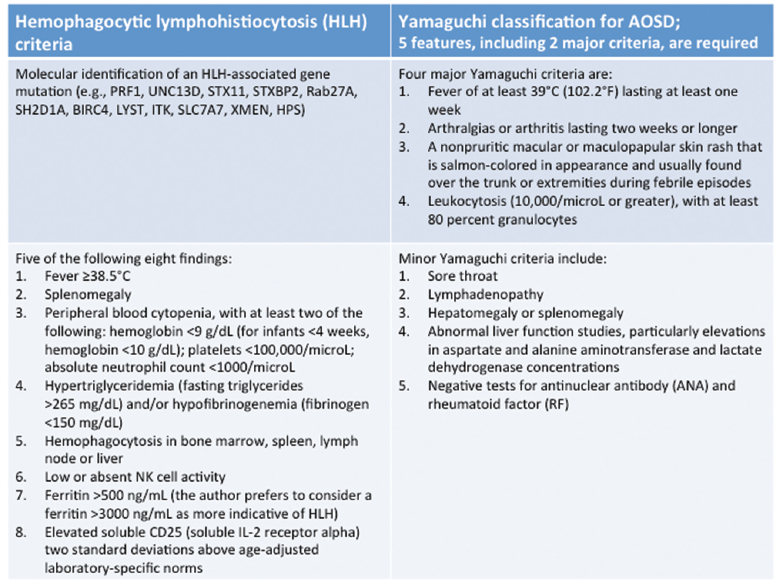
Reviewing the criteria for HLH, we noted that she met five out of the eight criteria, including fever, splenomegaly, pancytopenia, hypertriglyceridemia, a ferritin greater than 500, as well as hemophagocytosis in the bone marrow (see Table 1).2-4
The suspicion of HLH led to the question of etiology and whether it might be related to adult-onset Still’s disease (AOSD). She fulfilled the Yamaguchi criteria for AOSD, including two major criteria (fever and arthralgias lasting two or more weeks) as well as three minor criteria (splenomegaly, abnormal liver function studies with elevations and negative tests for ANA and RF) (see Table 1).2-4
Click for larger image
Table 1: HLH (left) & AOSD (right) Criteria
The patient remained febrile to 104ºF with abdominal pain, which had now progressed to involve her left lower quadrant and right upper quadrant with diffuse myalgias, fatigue and diffuse arthralgias in her hands. Given her ongoing symptoms, suspicion of HLH due to AOSD and otherwise negative evaluation, she was administered pulse doses of steroids 1 g IV solumedrol for three days followed by daily prednisone (1 mg/kg).