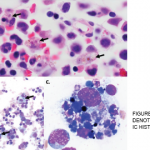
In another retrospective study including 162 patients diagnosed with HLH, 56% of cases were associated with a hematologic malignancy, especially non-Hodgkin’s lymphomas; only 70% of patients had features of hemophagocytosis in the bone marrow.8
An elevated sIL-2R two standard deviations above age-adjusted laboratory-specific norms is included in the diagnostic criteria for HLH. This is a marker for the presence of activated lymphocytes, which is usually measured by enzyme-linked immunosorbent assay.1 A normal level of sIL-2R for adults depends on the number of cells producing the IL-2 receptor and the number of molecules per cell. Although there is wide variation in the normal range of sIL-2R between laboratories, a value greater than two standard deviations above the normal range for a particular laboratory (or 2,400 U/mL in pediatric data sets9) has been found to be diagnostically useful.
A retrospective analysis of the laboratory findings of 21 patients with HLH (11 lymphoma-associated HLH and 10 benign disease-associated HLH) found the mean sIL-2R levels were significantly higher in the lymphoma-associated group (13,451 U/mL vs. 4,176 U/mL, P=0.0031), and ferritin levels were higher in the benign disease-associated HLH group (20,462 ng/mL vs. 2,561 ng/mL, P=0.0031).7 The mean serum sIL-2R:ferritin ratio of patients with lymphoma-associated disease was markedly higher than that of patients with benign disease-associated HLH (8.5 vs. 0.7, P=0.0004).7
This study suggests that the sIL‑2R:ferritin ratio can be useful as a marker in the diagnosis of LAHS.7
Treatment of HLH
The aim of therapy in patients with HLH is to suppress inflammation by destroying the responsible immune cells. Based on the HLH-94 protocol, treatment for acutely ill patients with primary HLH should begin with eight weeks of induction therapy with etoposide and dexamethasone, with intrathecal methotrexate therapy for any central nervous system involvement.
It is important to start therapy as soon as the diagnosis is recognized given the high mortality of untreated disease. Once induction is completed, patients are weaned off therapy while those patients who are not improving are continued on therapy until they can be offered allogeneic hematopoietic cell transplantation (HCT). Generally, stem cell transplant is needed for those patients with an HLH mutation, central nervous system disease or disease relapse.10 Median survival has been shown to be 54% at 6.2 years with treatment based on the HLH-94 protocol.10
In cases of secondary HLH, it’s essential to treat the underlying triggering condition, such as the infection, rheumatic disease or malignancy. Response to therapy is followed by monitoring the disease specific markers.
Conclusion
HLH is a life-threatening condition that requires prompt diagnosis and management. In patients with suspected HLH, more than one bone marrow biopsy may be needed for diagnosis, as in our patient. Patients with HLH should undergo thorough screening for malignancy, starting with age- and gender-