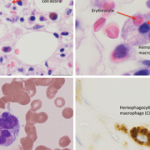
Unfortunately, the application of the HLH diagnostic criteria to systemic JIA patients with suspected MAS is problematic. Some of the HLH markers such as lymphadenopathy, splenomegaly, and hyperferritinemia are common features of active SJIA itself and, therefore, do not distinguish MAS from a conventional SJIA flare. Other HLH criteria, such as cytopenias and hypofibrinogenemia, become evident only at the late stages. This is related to the fact that systemic JIA patients often have increased white blood cell and platelet counts as well as increased serum levels of fibrinogen as a part of the inflammatory response seen in this disease. Therefore, when these patients develop MAS, they may only reach the degree of cytopenias and hypofibrinogenemia seen in HLH at the late stages of the syndrome when their management becomes challenging. The application of criteria is even more problematic for the diagnosis of MAS in patients with SLE in whom autoimmune cytopenias are common and difficult to distinguish from those caused by MAS. In these patients, the presence of extreme hyperferritinemia and lactate dehydrogenase elevation should raise suspicion for MAS. Attempts to modify the HLH criteria to increase their sensitivity and specificity for the diagnosis of MAS in rheumatic conditions have been initiated.19
Differential Diagnosis
In addition to distinguishing MAS from a flare of an underlying rheumatologic disease, one must consider in differential diagnosis other clinical entities associated with hepatic dysfunction, coagulopathy, cytopenias, or encephalopathy. In some MAS patients, the combination of hepatic dysfunction with encephalopathy may be reminiscent of Reye syndrome. The diagnosis of Reye syndrome, however, is based of the presence of a viral prodrome, unexplained vomiting, behavioral changes, and a distinctive chemical profile characterized by rapid coordinated increase in serum aminotransferase levels, blood ammonia, and prothrombin time with relatively minimal changes in serum bilirubin. The DIC-like coagulopathy seen in MAS is not a feature of Reye syndrome. Conversely, a sharp increase in blood ammonia levels, an important feature of Reye syndrome, is usually very mild in MAS. The hemorrhagic syndrome seen in MAS may resemble thrombotic thrombocytopenic purpura. However, microangiopathic anemia with emergence of fragmented red blood cells in peripheral circulation, a central feature of thrombotic thrombocytopenic purpura, is usually not seen in MAS. It is also important to differentiate MAS from malignancy-associated HLH and malignant histiocytic disorders. Some other important differential diagnoses include sepsis and drug reactions.