Results: A total of 289 patients were enrolled in the randomized cohort, and 365 patients were enrolled in the observational cohort. At two years, 67% of patients who were randomly assigned to surgery had undergone surgery, whereas 43% of those who were randomly assigned to receive nonsurgical care had also undergone surgery. Despite the high level of nonadherence, the intention-to-treat analysis of the randomized cohort showed a significant treatment effect favoring surgery on the SF-36 scale for bodily pain, with a mean difference in change from baseline of 7.8 (95% confidence interval, 1.5 to 14.1); however, there was no significant difference in scores of physical function or on the Oswestry Disability Index. The as-treated analysis, which combined both cohorts and was adjusted for potential confounders, showed a significant advantage for surgery by three months for all primary outcomes; these changes remained significant at two years.
Conclusions: In the combined as-treated analysis, patients who underwent surgery showed significantly more improvement in all primary outcomes than did patients who were treated nonsurgically.
Commentary
In 2005, the Cochrane Review group reported the lack of clear or decisive data concerning the efficacy of surgical decompression for treating spinal stenosis. Although clinical studies on surgical therapy have been published, they have been limited by, among other things, the inclusion of patients with spinal instability (spondylolisthesis) who required fusion as a component of surgical therapy. The studies included small groups of patients without significant improvement from surgical intervention. In response to paucity of clinical trials studying an adequate number of similar stenosis subjects, the Spine Patient Outcomes Research Trial (SPORT) group completed a clinical trial investigating the relative benefit of surgical versus nonsurgical therapy for patients with spinal stenosis without spondylolisthesis.
The conclusion of the Weinstein et al. article is that surgical decompression offers greater improvement in pain and physical function than nonsurgical therapy. Have the authors, however, identified those individuals who are good surgical candidates and have they demonstrated a “good” outcome for these spinal stenosis sufferers? The study had 654 individuals: 400 surgical and 254 nonsurgical participants. The individuals eligible to participate in this trial had neurogenic claudication for a minimum of three months and radiographic evidence of spinal stenosis at one or more levels without instability; they were also judged to be surgical candidates although this criterion was never fully defined.
The authors describe the group who underwent surgery as younger, working, with more pain, lower level of function, psychological distress, more severe stenosis, and with worsening symptoms. A slim majority of participants had symptom duration of more than six months, but the scope of the persistence of radicular pain was not mentioned. An incongruity arises when matching duration of symptoms with the radiographic grading of stenotic severity. A majority of individuals are described with evidence of severe stenosis at one or more levels. I would have expected these individuals to have been symptomatic for a longer period of time than six months.
The most frequent comorbidity in these individuals with an average age of 65 was the presence of other joint disease. The impact of this finding on physical function is not mentioned. Nonsurgical therapy was left to the discretion of the practitioner who was most likely an orthopedic surgeon. The nonsurgical candidates may not have been individuals who failed medical therapy, but those who never received a full complement of educational, pharmaceutical, physical, and injection interventions.
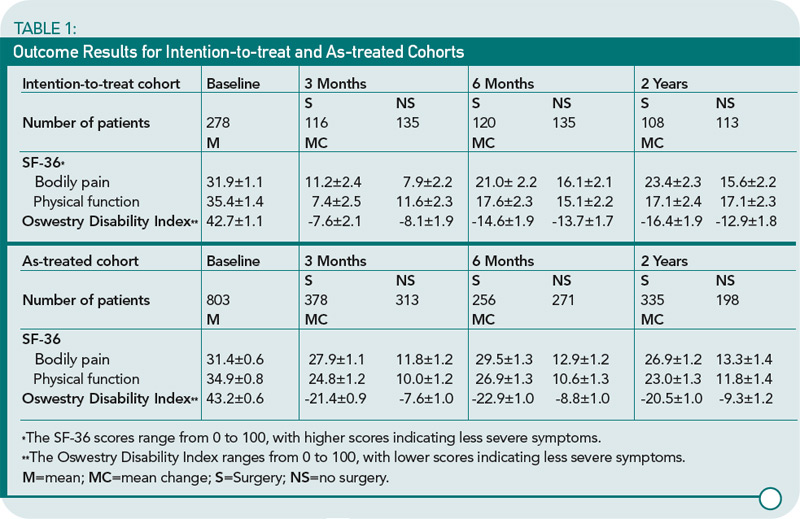
The study design included two groups: 1) a randomized cohort assigned to one therapy (n=289) or 2) an observational cohort who self-selected therapy (n=365). A major complicating factor of this study was that 33% of surgical patients who chose medical therapy, and 43% of nonsurgical patients who underwent laminectomy. The authors completed both an intention-to-treat and as-treated analysis because of the frequent crossovers. A partial summary is included in Table 1 (below).
At two years, in the intention-to-treat analysis, surgical patients had a significant improvement in pain but not in physical function or disability. In the as-treated analysis, however, the superiority of surgical decompression over nonsurgical therapy was shown in pain and physical improvement. The as-treated group included individuals who chose the therapy that they thought would help them the most in the setting of their pain severity and physical dysfunction. I believe that the general applicability of the conclusions of the study is diluted both by the very specific clinical characteristics of the study subjects and the absence of randomization for a significant proportion of patients. The Cochrane Review group will probably say that more randomized studies are needed, despite the addition of this clinical trial to the medical literature.
So what are busy clinicians going to recommend to their spinal stenosis patients?1 I think clinicians need to explain the nonsurgical and surgical options to patients. Patients who are younger, with no associated joint disease, with single-level stenosis, and without spondylolisthesis who want surgery have a good opportunity to improve with surgical intervention. Patients who chose medical therapy may be more symptomatic for a period of time, but will become better over the subsequent two years.
Reference
IMMUNOLOGY
Potential New Model to study immunological disturbances
By Maripat Corr, MD
Yang YG, Lindahl T, Barnes DE. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell. 2007;131:873-886.
Abstract
Trex1 is the major 3’ DNA exonuclease in mammalian cells, and mutations in the human TREX1 gene can cause Aicardi-Goutières syndrome, characterized by perturbed immunity. Similarly, Trex1(-/-) mice have an autoinflammatory phenotype; however, the mechanism of Trex1-deficient disease is unknown. We report that Trex1, ordinarily associated with the endoplasmic reticulum (ER), relocalizes to the S phase nucleus after gamma irradiation or hydroxyurea treatment. Notably, Trex1-deficient cells show defective G1/S transition and chronic ATM-dependent checkpoint activation, even in the absence of exogenous stress, correlating with persistent single-stranded DNA molecules produced in S phase, which accumulate in the ER. Our data indicate that Trex1 acts on a single-stranded DNA polynucleotide species generated from processing of aberrant replication intermediates to attenuate DNA damage checkpoint signaling and prevent pathological immune activation.
Commentary
In 1984, two French pediatric neurologists—Jean Aicardi and Francoise Goutières—described, in a Portuguese family, a rare neurological condition that is attracting attention as a model to study immunological disturbances in autoimmunity. This condition, Aicardi-Goutières syndrome (AGS), shows autosomal recessive inheritance and is characterized by encephalopathy with elevated lymphocytes in the cerebrospinal fluid, calcification of the basal ganglia, and white matter demyelination.1 In infancy, AGS typically manifests as progressive microcephaly, spasticity, dystonia, and psychomotor retardation. However, AGS is a heterogenous disorder and phenotypic variability includes patients with apparently static or slowly progressive disease, sometimes presenting after several months of normal development.
Recently, mutations in TREX1 and in the genes encoding the three nonallelic components of the RNASEH2 protein complex have been identified in AGS.2-4 TREX1 encodes the major 3’->5’ DNA repair exonuclease in mammalian cells, DNase III/TREX1. Several monoallelic mutations in TREX1 have recently been described in individuals with systemic lupus erythematosus and familial chilblain lupus.5 Despite these known clinical associations, the mechanisms by which TREX1-deficiency promotes disease have remained obscure. In an important new study, Yang et al. describe the accumulation of cytoplasmic single stranded (ss) DNA that is associated with this defect in cell cycle modulation, which could provide a link between this deficiency and chronic inflammation.
The investigative group observed that the protein TREX1 can translocate from the ER in the cytoplasm to the nucleus at the time of DNA synthesis for replication. Fibroblasts from TREX1-null mice did not progress normally through the cell cycle, however, and proliferated slowly. Furthermore, TREX1 deficient cells failed to arrest properly in S and G2/M-phases of the cell cycle following DNA damage with irradiation. Molecular dissection of the checkpoints in the cell cycle associated with DNA damage repair indicated that the TREX1-deficient cells exhibited chronic activity of the ataxia-telangiectasia mutated (ATM) kinase, resulting in lower CHK2 levels and increased levels of active p53 and p21.
As TREX1 was previously described to use ssDNA as a substrate, the researchers hypothesized that loss of TREX1 activity could result in ssDNA accumulation. Indeed, TREX1-null fibroblasts exhibited a marked increase in the amount of ssDNA polynucleotides between 60 and 65 bases. Surprisingly, the accumulation was not in the nucleus, but in the cytoplasm. Using fluorescent microscopy they demonstrated—by co-localization with an ER-specific marker, calreticulin, that the ssDNA is associated with the ER. Experiments with primary fibroblasts from AGS patients with homozygous mutations in Trex1 demonstrated a similar cell cycle profile as the murine cells and an increase in ER-associated ssDNA.
From these studies, it appears that TREX1-deficiency results in chronic activation of the ATM kinase–dependent checkpoint and the accumulation of ssDNA oligonucleoides within the cytoplasm of replicating cells. Despite the aberrant cell cycle and checkpoint activity, mice that lack TREX1 and AGS-affected individuals do not have an increased tumor incidence. Rather, the hallmark of AGS is the presence of elevated levels of interferon-α in the CSF, mimicking congenital infections (pseudo-TORCH syndrome). Aberrant immune activation is also indicated by the chilblain lesions and the small number of AGS-affected children who develop antinuclear antibodies, hypothyroidism, and type I diabetes.3 Innate immune receptors, including the Toll-like receptors, recognize strands of nucleic acids as ligands, but have not been identified as a specific pathway for the interferon-α production in the pathogenesis of AGS.6
An alternative mechanism for the effects of TREX deficiency on autoimmunity concerns its role in cell death. TREX1 activity has been implicated in a specific caspase-independent cell-death pathway utilized by cytotoxic T lymphocytes (CTL) and natural killer (NK) cells during antiviral responses.7 Type I interferons stimulate the activity of these cells. In turn they release granzymes and perforin to induce death in target cells. Defective granzyme A activity as a result of Trex1 deficiency could result in the inability to eliminate autoreactive lymphocytes. Alternatively, genetic defects in perforin have resulted in sustained cytokine production by NK cells and CTLs, which may occur in AGS.8 The identification of the malfunctioning proteins in AGS provides evidence that a defect in DNA processing can lead to chronic expression of inflammatory cytokines, including interferon-α and autoimmunity. As often occurs in science, insights into disease can come from unexpected places and it is intriguing that a study of a relatively rare neurological disease may enhance our understanding of more global mechanisms underlying autoimmunity.
References
- Aicardi J, Goutières F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol. 1984;15:49-54.
- Crow YJ, Hayward BE, Parmar R, et al. Mutations in the gene encoding the 3’-5’ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat Genet. 2006;38:917-920.
- Rice G, Patrick T, Parmar R, et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am J Hum Genet. 2007;81:713-725.
- Crow YJ, Leitch A, Hayward BE, et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutières syndrome and mimic congenital viral brain infection. Nat Genet. 2006;38:910-916.
- Lee-Kirsch MA, Gong M, Chowdhury D, et al. Mutations in the gene encoding the 3’-5’ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. 2007;39:1065-1067.
- Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7:131-137.
- Chowdhury D, Beresford PJ, Zhu P, et al. The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol Cell. 2006;23:133-142.
- Marcenaro S, Gallo F, Martini S, et al. Analysis of natural killer-cell function in familial hemophagocytic lymphohistiocytosis (FHL). Blood. 2006;108:2316-2323.
