E.G.L. Bywaters, the first to describe the syndrome in adults, was an Englishman trained in rheumatology by the legendary Walter Bauer, MD. at the Massachusetts General Hospital. When World War II broke out, Dr. Bywaters returned to England. During the bombing of London, he discovered that renal failure in crush syndrome resulted from the release of myoglobin. In 1971, he went on to describe 14 adults with a presentation similar to that described by Still, and this syndrome came to be known as adult Still’s disease (AOSD).
A daily (quotidian) fever is characteristic of AOSD and is sometimes described as a picket fence fever, spiking in the afternoon or early evening, then returning to normal. This occurs in up to 80% of AOSD patients, tends to last around four hours and often precedes the development of other manifestations. Severe supparative pharyngitis sometimes precedes fever or rash.
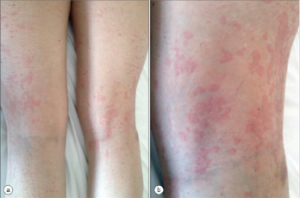
Figure 1
AOSD Rash
The rash also has characteristic features: macular or maculopapular, nonpruritic, evanescent, and of a faint salmon or pinkish color vessels (see Figure 1). It tends to occur with the fever and may be brought on by a hot shower. (My mentor called it the intern’s rash, because the intern was the only doctor in the hospital in the evening when the rash occurs.) The rash exhibits the Koebner phenomenon, which means that it can be brought on by mild trauma. (Heinrich Koebner, MD, described this in psoriasis patients. He was better known for inoculating his arms with three different fungi to demonstrate the infectious nature of the rashes they caused). Stroking the skin with the finger will sometimes provoke the rash (I like to draw an S), and examination of the upper back will often reveal pink lines that correspond to the creases in the bed sheets. Pathologically, the findings are nondiagnostic: dermal edema, perivascular inflammation in the superficial dermis with lymphocytes and histiocytes, with C3 deposition in the blood.