Currently Available Biosimilars—And Their Complications
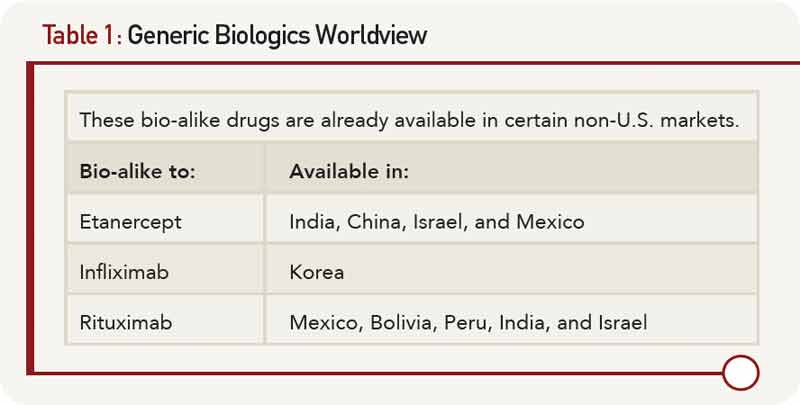
Biosimilars for human growth factors and hormones have already been developed and brought to market in the E.U. These include epoietin, insulin, filgrastim, and somatatropin. These are complex molecules; for example, somatotropin has a molecular weight that is 100 times greater than aspirin. The advent of a less costly biosimilar, filgrastim (cf. Neupogen), resulted in a dramatic increase in the use of this agent in the U.K. On the other hand, the development of the erythropoietin biosimilar has faced many challenges. Seemingly small changes in manufacturing have led to dramatic consequences. For example, the tungsten-containing component of the rubber plunger that was used in the prefilled syringe resulted in unfolding of the protein. This change in structure led to the formation of antibodies to the biosimilar that cross-reacted with native erythropoietin. As a result, between 1998 and 2004, there were 175 cases of pure red-cell aplasia in patients taking the drug. The newly approved erythropoietin biosimilar (Omontys) was recalled due to patients developing allergic reactions, some of which were fatal.