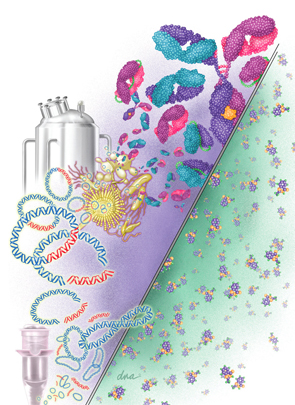
Biosimilars are much bigger and more complex on the molecular level than small-
molecule drugs. On the right is the small-molecule structure of aspirin (21 molecules). On the left is a monoclonal antibody made up of 10,000–20,000 molecules. The various biosimilar drugs depicted include monoclonal antibodies, scFv, Fab, and scFab, whereas the small-molecule drugs are identical. Also shown is a vial used for recombinant DNA procedures with plasmids as vectors and the desired fragments of DNA that are attached to the plasmids, forming recombinant plasmids. These are inserted into various living organisms, which produce the desired biosimilars in a fermentation tank.
DNA Illustrations/ScienceSource.com
On Feb. 9, the Arthritis Advisory Committee of the U.S. Food and Drug Administration (FDA) met in Washington, D.C., and recommended the approval of CT-P13, a proposed biosimilar to infliximab (Remicade), for indications including the treatment of rheumatoid arthritis, in combination with methotrexate; ankylosing spondylitis; and psoriatic arthritis. The human monoclonal antibody would be the first biosimilar available in the U.S. market to treat rheumatic diseases.