A cytotoxic or antiproliferative effect of methotrexate is clearly consistent with its known actions in the treatment of malignancies; however, methotrexate-mediated cytotoxicity cannot fully explain the antiinflammatory actions of methotrexate. Perhaps the most telling point in this regard is that, unlike the antiproliferative effects of methotrexate, neither folic acid nor folinic acid (except when given at the same time as methotrexate) consistently reduces or reverses the antiinflammatory effects of methotrexate in RA in randomized, blinded prospective trials.1 Moreover, the development of leukopenia, common in patients receiving high doses of methotrexate to treat malignancies, is a sign to cut back the dose or stop the use of methotrexate in treating patients with RA. Thus, other explanations for methotrexate’s mechanism of action have been explored (see Figure 1).
Figure 1
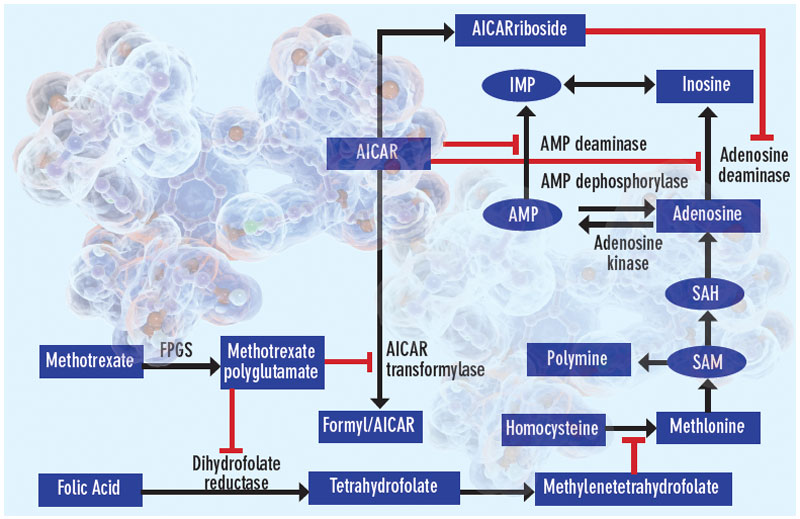
Proposed mechanisms of the antiinflammatory actions of methotrexate. Methotrexate exerts its antiinflammatory actions through a number of cellular mechanisms. Competitive inhibition of dihydrofolate reductase diminishes the de novo synthesis of purines and pyrimidines by preventing the regeneration from dihydrofolate of tetrahydrofolate, which is essential for the generation of folate cofactors required for purine and pyrimidine synthesis. Reduction of the levels of methyl donors, such as tetrahydrofolate and methyltetrahydrofolate, by the inhibition of dihydrofolate reductase results in the inhibition of the generation of lymphotoxic polyamines through methionine and SAM. Inhibition of AiCAr transformylase results in an increase in intracellular AiCAr levels. This increase has potent inhibitory effects on AMP deaminase and adenosine deaminase, which are involved in the catabolism of AMP and adenosine to IMP and inosine, respectively. The consequent accumulation of adenosine confers antiinflammatory effects. Levels of AiCArriboside, a metabolite of AiCAr, also accumulate and inhibit adenosine deaminase.