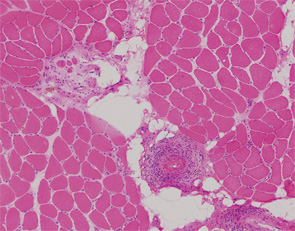
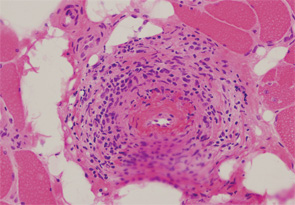
Patients with diffuse systemic sclerosis (dSS) with an associated ANCA-positive vasculitis have been infrequently described. In this article, we report the case of a 53-year-old woman, previously diagnosed with dSS, who developed an ANCA-associated vasculitis, which manifest as peripheral nerve and muscle disease.
Scleroderma is a disease characterized by sclerosis of the skin and visceral organs, vasculopathy, Raynaud’s phenomenon and the presence of autoantibodies. The disease spectrum is wide, with systemic and localized forms, depending on the extent of skin thickening. There is also a vasculopathy involving multiple organ systems that leads to the presence of cutaneous and mucosal telangiectasias, digital ulcers and tissue ischemia. Medium-size vessels can also be involved, sometimes leading to scleroderma renal crisis or pulmonary artery hypertension. However, scleroderma is rarely associated with an ANCA-positive vasculitis. We report this unique situation.
The development of vasculitis in patients with systemic sclerosis is highly uncommon.
Initial laboratory tests included a normocytic and normochromic anemia, thrombocytosis, elevated acute-phase reactants and normal serum creatinine. MPO-ANCA titer was 174 AU (normal <10 UI/mL). PR-3-ANCA was negative. Antiglomerular basal membrane and anti-Jo-1 antibodies were negative, and complement C3 and C4 were within normal limits. An electromyogram showed evidence of a moderate myopathy without polyneuropathy or myositis. A muscle and peripheral nerve biopsy demonstrated features of a necrotizing vasculitis with signs of axonal degeneration and severe denervation attributable to ischemic neuropathy (see figures, right).
Therapy was initiated with a high dose of prednisolone (bolus of 1 mg/kg/day IV for three days, followed by 60 mg PO daily) and cyclophosphamide treatment (five monthly IV boluses of 750 mg each) was initiated. A few weeks later, maintenance immunosuppressive therapy was introduced with azathioprine (150 mg day PO), and her hypoesthesia and weakness gradually disappeared.
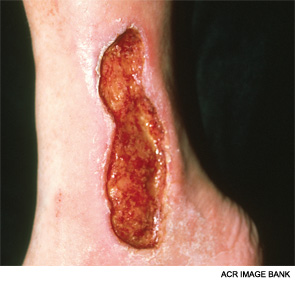
She improved progressively and currently has no peripheral neuropathy or myopathy. Her pulmonary involvement has been stable. However, over time, there has been more extensive cutaneous sclerosis and the development of more digital ulcers.
Discussion
The development of vasculitis in patients with SS is highly uncommon.1 Whereas the histopathology of SS vasculopathy demonstrates a myofibroblast proliferation and matrix deposition in the subendothelial layer leading to occlusion, the histopathology of vasculitis is characterized by the presence of mononuclear infiltrates and destruction of the vessel wall.
A recent publication by the French Vasculitis Study Group identified nine patients with SS and vasculitis.2 Another study described the characteristics of a series of 40 patients with SS and associated systemic vasculitis, finding that the main clinical manifestation was a rapidly progressive glomerulonephritis (83%), followed by alveolar hemorrhage (28%), limb ischemia (13%) and skin vasculitis (10%).3
The presence of a positive ANCA in patients with SS is estimated to be in the range of 7–13%, but its clinical relevance is unknown. MPO-ANCA is more frequent than PR3-ANCA in patients with SS.4
Conclusion
The patient improved after immunosuppressive therapy. She has not experienced a new outbreak of vasculitis, although her cutaneous disease related to scleroderma has progressed.
Carmen Busca Arenzana is a medical resident in the internal medicine department of the Hospital La Paz in Madrid, Spain.
Ander Mayor-Ibarguren is a medical resident in the dermatology department of the Hospital La Paz in Madrid, Spain.
Amara Pieren-Salazar is a medical resident in the rheumatology department of the Hospital La Paz in Madrid, Spain.
Natalia Martin-Suñe, MD, works in the internal medicine department of the Hospital La Paz in Madrid, Spain.
Isabel Esteban-Rodríguez, MD, works in the pathology department of the Hospital La Paz in Madrid. Spain.
Juan José Ríos-Blanco, MD, works in the internal medicine department of the Hospital La Paz in Madrid, Spain.
Francisco Arnalich-Fernandez, MD, works in the internal medicine department of the Hospital La Paz in Madrid, Spain.
References
- Quéméneur T, Mouthon L, Cacoub P, et al. Systemic vasculitis during the course of systemic sclerosis. Medicine (Baltimore). 2013 Jan;92(1):1–9.
- Arad U, Balbir-Gurman A, Doenyas-Barak K, et al. Anti-neutrophil associated vasculitis in systemic sclerosis. Semin Arthritis Rheum. 2011 Oct;41(2):223–229.
- Rho YH, Choi SJ, Lee YH, et al. Scleroderma associated with ANCA-associated vasculitis. Rheumatol Int. 2006 Mar;26(5):369–375.
- Mouthon L, Millet A, Régent A, et al. Physiopathologie des vascularites ANCA-positives. Presse Med. 2012 Oct;41(10):996–1003.
Acknowledgments: The authors would like to thank Martin J. Smyth for his help in correcting their English.



