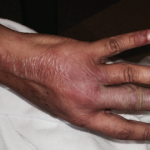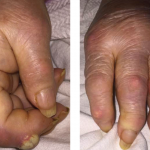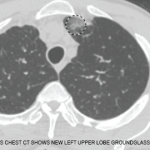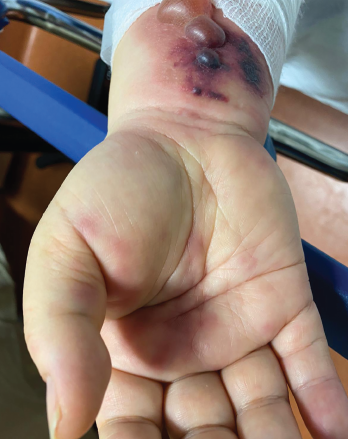
Figure 1. Tense hemorrhagic bullae developed over the course of the patient’s disease. Tender purpuric papules on the palms and marked hand swelling are also apparent.
Eosinophilic granulomatosis with polyangiitis (EGPA) is an anti-neutrophil cytoplasmic antibody-associated vasculitis typically characterized by asthma, peripheral eosinophilia and medium- to small-vessel necrotizing vasculitis. Cutaneous manifestations in EGPA are diverse. Palpable purpura is the most common presentation, but urticaria, erythematous macules and papules, livedo reticularis, digital necrosis and cutaneous nodules have also been described.1 Non-hemorrhagic bullae are a rare initial manifestation of EGPA.2-5
Case Presentation
A 58-year-old woman with a history of adult-onset asthma and sinusitis presented with an acute bullous skin eruption. She had been prescribed amoxicillin-clavulanate for ear pain; however, after five days of antibiotic therapy, she developed pruritus overlying her chest and neck. Over the next few days, she developed diffuse extensive painful bullae and erythematous plaques.