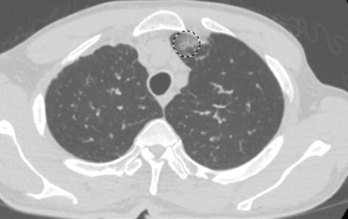
Figure 1. This chest CT shows new left upper lobe groundglass opacity.
Eosinophilic granulomatosis with polyangiitis (EGPA), also known as Churg-Strauss syndrome or allergic granulomatosis and angiitis, is a rare small- and medium-vessel vasculitis. This disease was first described by American pathologists Jacob Churg and Lotte Strauss in 1951.1
Although the vasculitis is often not apparent in the initial phases of the disease, EGPA can affect any organ system. It often manifests as peripheral blood eosinophilia, asthma and/or allergic rhinitis. In addition, patients frequently experience non-specific systemic findings, such as fever, weight loss, malaise, myalgias and arthralgias. Classified as an anti-neutrophil cytoplasmic antibody (ANCA) associated vasculitis, along with granulomatosis with polyangiitis and microscopic polyangiitis, EGPA is the least common of the three.2
The Case
A 63-year-old man with a past medical history of prostate cancer (treated with radical prostatectomy), hyperlipidemia and asthma presented to our emergency department with a one-day history of worsening shortness of breath and dyspnea on exertion. The patient did not have any previous personal or family history of rheumatologic disease. Of note, he was diagnosed with asthma within one year of his initial presentation.
Two months earlier, the patient was evaluated by his primary care physician for watery diarrhea. His diarrhea had resolved, but over the course of the next two weeks, the patient noted worsening lower extremity numbness and tingling. Labs at that time revealed leukocytosis of 30.9×103/µL (50.3% eosinophils).
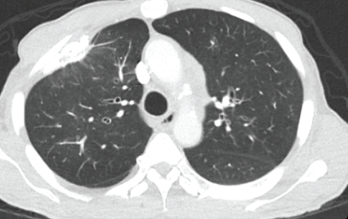
Figure 2. This chest CT shows improved aeration of the left upper lobe compared with Figure 1.
The patient’s complete blood count one year earlier had revealed a normal white blood cell (WBC) count with 5.1% eosinophils. The patient was hospitalized briefly for worsening leukocytosis and eosinophilia in the setting of thrombocytosis. He underwent extensive workups with hematology/oncology and infectious disease experts, all of which proved unremarkable. A computed tomography (CT) scan of his chest revealed densely calcified pleural plaques involving the anterior aspect of the lung’s right upper lobe with an adjacent parenchymal opacity. He refused further inpatient workup and was discharged on oral antibiotics and an oral prednisone taper.
In the month preceding his presentation at our emergency department, the patient underwent further outpatient workup. A repeat chest CT demonstrated a new left upper lobe groundglass opacity with new mediastinal lymphadenopathy in addition to stable, upper right lobe, partially calcified, pleural thickening (see Figure 1). The patient continued to experience worsening paresthesias of his feet bilaterally (the left more so than the right), and an autoimmune workup (see Table 1) demonstrated elevated inflammation markers. A bone marrow biopsy revealed reactive eosinophilia without evidence of underlying leukemia or lymphoproliferative process.
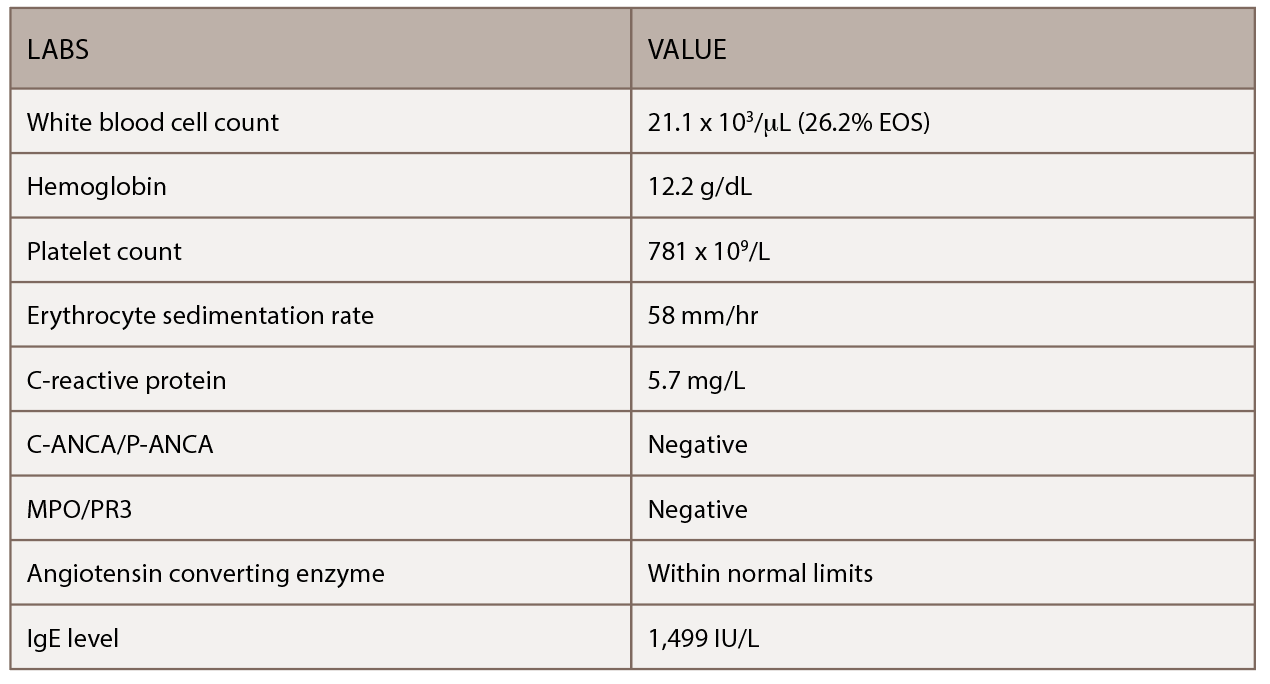
(click for larger image) Table 1: This table shows the patient’s initial outpatient rheumatology serologic workup.
In our emergency department, the patient was immediately evaluated for worsening shortness of breath and wheezing, which were unresponsive to albuterol. He did not have a fever and denied any weight loss, night sweats, headaches, visual changes, rash or joint pain. The patient had diarrhea and distal lower extremity weakness with numbness and tingling of his feet (more so on the left than the right). His vital signs were stable, and labs revealed mild anemia, leukocytosis of 32,000/µL (upper limit of normal [ULN]: 11,000/µL), 39.4% eosinophils (ULN: 3%), thrombocytosis at 791,000/µL (ULN: 450,000/µL) and an elevated paraprotein gap (total protein-albumin >4).
An electrocardiogram (ECG) proved unremarkable. Serial cardiac enzymes demonstrated evidence of myocardial injury (first set: troponin 0.95 units, which increased to 2.1 units ng/mL). The ECG demonstrated a normal ejection fraction without diastolic dysfunction in the setting of a pericardial effusion and a mildly dilated inferior vena cava. Cardiac catheterization did not show any evidence of obstructive coronary artery disease. The patient was clinically diagnosed with myocarditis and was given aspirin, atorvastatin and metoprolol.
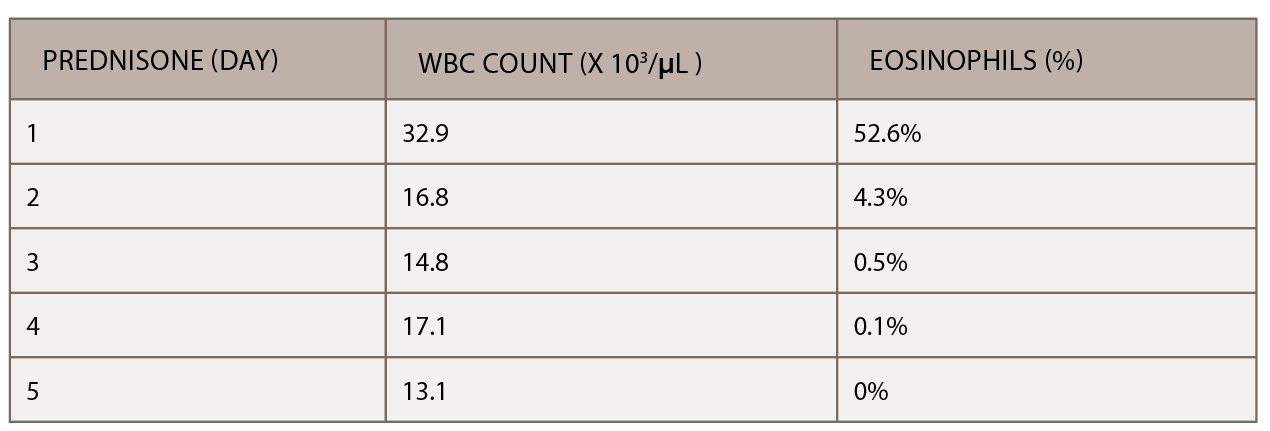
(click for larger image) Table 2: The initiation of inpatient prednisone produced a down-trending white blood cell count and normalization of percentage of eosinophils.
A chest CT demonstrated a persistent parenchymal opacity in the right upper lobe, with improved left upper lobe aeration compared with the prior left upper lobe opacity (see Figure 2). A sinus CT demonstrated severe sinonasal mucosal disease with near complete opacification of the bilateral frontal and ethmoid sinuses (see Figures 3 and 4). Inpatient labs revealed elevated inflammatory markers and a markedly elevated rheumatoid factor (RF) of greater than 600 IU/mL. With concern for underlying systemic vasculitis, we initiated prednisone at 1 mg/kg/day, and subsequent labs demonstrated a drop in white blood cells and normalization of his eosinophil percentage (see Table 2). The patient noted subjective improvement in his wheezing, diarrhea and motor strength.
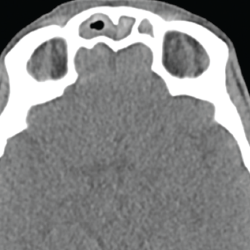
Figure 3. This sinus CT shows severe nasomucosal disease with near complete opacification of the bilateral frontal sinuses.
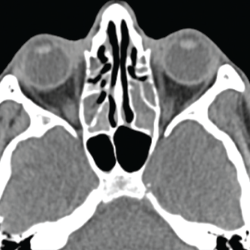
Figure 4. This sinus CT shows severe nasomucosal disease with near complete opacification of the bilateral ethmoid sinuses.
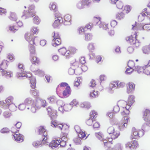
An esophagogastroduodenoscopy (EGD) and colonoscopy demonstrated chronic gastritis and patchy areas of severely congested, erythematous and friable mucosa throughout the entire colon. A bronchoscopy with bronchoalveolar lavage and a transbronchial lung biopsy showed bronchiole walls with a reticular basement membrane containing scattered infiltrates of eosinophils in the adventitia (see Figure 5). Fine needle aspiration of lymph nodes ruled out epithelial malignancy. Before the EGD and colonoscopy, active gastrointestinal bleeding was ruled out, and the decision was made to administer a reduced pulse dose of intravenous methylprednisolone at 500 mg/day for three days to hasten neurologic recovery.
Prior to discharge, the patient underwent a left sural nerve biopsy, which showed vasculitic neuropathy with endoneurial fibrosis. Pathology demonstrated arteries in longitudinal sections with thrombosis and eosinophils mixed with macrophages and T cells, consistent with vasculitis (see Figure 6). The patient experienced mild improvement of his neuropathy and was subsequently discharged on prednisone 1 mg/kg/day with outpatient rheumatology follow-up.
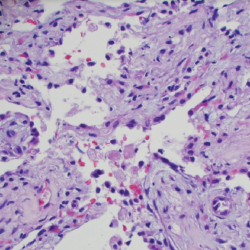
Figure 5. A transbronchial lung biopsy showed bronchiole walls containing scattered infiltrates of eosinophils in the adventitia.
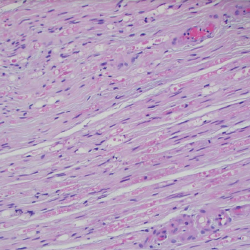
Figure 6. A sural nerve biopsy demonstrated arteries in longitudinal sections with thrombosis and eosinophilic infiltrate, consistent with vasculitis.
Discussion
EGPA was first described more than 60 years ago, but its underlying mechanisms remain poorly understood. The 1990 ACR classification criteria for EGPA include: 1) asthma, 2) eosinophilia (>10% of total white cell count), 3) neuropathy, 4) non-fixed pulmonary infiltrates, 5) paranasal sinus abnormality and 6) extravascular eosinophil infiltration on biopsy.6 For classification purposes, a patient is said to have EGPA if at least four of these six criteria are present (sensitivity of 85%; specificity of 99.7%).6
Outside the ACR criteria, additional classification criteria exist for EGPA. Per the 1984 Lanham classification criteria, a patient has a diagnosis of EGPA if all three of the following criteria are present: 1) asthma, 2) eosinophilia (>10% WBC count or >1.5×109) and 3) systemic vasculitis affecting at least two extrapulmonary sites.7
The 2012 Revised International Chapel Hill Consensus Conference nomenclature of vasculitides, note EGPA is an “eosinophil-rich and necrotizing granulomatous inflammation often involving the respiratory tract, and necrotizing vasculitis predominantly affecting small to medium vessels with associated asthma and eosinophilia.”7
The recently published MIRRA Study (Investigating Mepolizumab in the Treatment of EGPA) defined EGPA as the presence of asthma and eosinophilia (>1.0×10^9/L and/or >10% of leukocytes) plus at least two of the following features: a biopsy showing histopathological evidence of eosinophilic vasculitis; perivascular eosinophilic infiltration; eosinophil-rich granulomatous inflammation; neuropathy (mono- or poly- with motor deficit or nerve conduction abnormality); pulmonary infiltrates (non-fixed); sino-nasal abnormality; cardiomyopathy (established by echo or MRI); glomerulonephritis (hematuria, red cell casts, proteinuria); alveolar hemorrhage (via bronchoalveolar lavage); palpable purpura; or being ANCA positive (myeloperoxidase or proteinase 3).8
No laboratory tests specific for EGPA exist, although eosinophilia is characteristic. ANCA are found in roughly 30–60% of EGPA patients.9 Our patient was ANCA negative. Myocarditis (as well as alveolar hemorrhage) more typically appears in ANCA-negative EGPA, as demonstrated in our case.10
The majority of ANCA-positive EGPA patients (70–75%) have antibodies directed against myeloperoxidase (MPO) with a perinuclear staining pattern (P-ANCA).11 Labs may also reveal leukocytosis, reactive thrombocytosis and a positive rheumatoid factor, all of which appeared in our patient.12 Imaging studies in EGPA can reveal transient and patchy opacities in up to 75% of patients, which was observed in our patient.13
Clinicians must understand the systemic nature of EGPA. Our patient had suspected multi-organ involvement, including upper airway/nasal, lower airway/pulmonary, gastrointestinal, cardiovascular and neurologic systems. Cardiac involvement is one of the more serious manifestations of EGPA, because clinical manifestations can include signs of heart failure, myocarditis, pericarditis and cardiac rhythm abnormalities.14,15
The main diseases to consider in the differential diagnosis of EGPA include hypereosinophilic syndrome, aspirin-exacerbated respiratory disease, the eosinophilic pneumonias and allergic bronchopulmonary aspergillosis, as well as the other two ANCA-associated vasculitides (i.e., granulomatosis with polyangiitis and microscopic polyangiitis). Treatment for patients with EGPA and evidence of systemic vasculitis includes prednisone (0.5–1 mg/kg/day).16 Cyclophosphamide is typically used in combination with steroids for severe, multi-organ disease.17 Maintenance and glucocorticoid sparing therapy includes azathioprine, methotrexate, leflunomide and a humanized monoclonal antibody to interleukin 5 (mepolizumab).18
Our case demonstrates a unique presentation of EGPA presenting with myocarditis before diagnosis. Although EGPA remains rare, consider it in patients presenting with myopericarditis. This may aid in early diagnosis and prompt immunosuppressive therapy to prevent long-term complications.
Ambreesh Chawla, MD, is a first-year rheumatology fellow at the University of Central Florida College of Medicine, Orlando, Fla. His clinical interests include vasculitis and connective tissue disease-related ILD.
Ashwini Komarla, MD, is a rheumatologist at the Orlando VA Medical Center. She is an assistant program director for the UCF/VA Rheumatology Fellowship Program.
Sujatha Vuyyuru, MD, is the department chief of rheumatology at the Orlando VA Medical Center. She is the program director for the UCF/VA Rheumatology Fellowship Program.
References
- Churg J, Strauss L. Allergic granulomatosis, allergic angiitis, and periarteritis nodosa. Am J Pathol. 1951 Mar-Apr;27(2):277–301.
- Keough KA, Specks U. Churg-Strauss syndrome. Semin Respir Crit Care Med. 2006 Apr;27(2):148–157.
- Mayo Clinic. Churg-Strauss syndrome—Symptoms.
- Churg A. Pulmonary angiitis and granulomatosis revisited. Hum Pathol. 1983 Oct;14(10):868–883.
- Conron M, Beynon HL. Churg-Strauss syndrome. Thorax. 2000 Oct;55(10):870–877.
- Masi AT, Hunder GG, Lie JT, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum. 1990 Aug;33(8):10941–100.
- Giofreddi A, Maritati F, Olivia E, et al. Eosinophilic granulomatosis with polyangiitis: An overview. Front Immunol. 2014 Nov 3;5:549.
- Wechsler ME, Akuthota P, Jayne D, et al. Mepolizumab or placebo for eosinophilic granulomatosis with polyangiitis. N Engl J Med. 2017 May 18;376(20):1921–1932.
- Cottin V, Bel E, Bottero P, et al. Revisiting the systemic vasculitis in eosinophilic granulomatosis with polyangiitis (Churg-Strauss): A study of 157 patients by the Groupe d’Etudes et de Recherche sur les Maladies Orphelines Pulmonaires and the European Respiratory Society Taskforce on eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Autoimmun Rev. 2017 Jan;16(1):1–9.
- Sinico RA, Di Toma L, Maggiore U, et al. Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg‐Strauss syndrome. Arthritis Rheum. 2005 Sep;52(9):2926–2935.
- Moosig F, Bremer JP, Hellmich B, et al. A vasculitis centre based management strategy leads to improved outcome in eosinophilic granulomatosis and polyangiitis (Churg-Strauss, EGPA): Monocentric experiences in 150 patients. Ann Rheum Dis. 2013 Jun;72(6):1011–1017.
- Guillevin L, Cohen P, Gayraud M, et al. Churg-Strauss syndrome. Clinical study and long-term follow-up of 96 patients. Medicine (Baltimore). 1999 Jan;78(1):26–37.
- Choi YH, Im JG, Han BK, et al. Thoracic manifestations of Churg-Strauss syndrome: Radiological and clinical findings. Chest. 2000 Jan;117(1):117–124.
- Neumann T, Manger B, Schmid M, et al. Cardiac involvement in Churg-Strauss syndrome: Impact of endomyocarditis. Medicine (Baltimore). 2009 Jul;88(4):236–243.
- Corradi D, Maestri R, Facchetti F. Postpartum Churg-Strauss syndrome with severe cardiac involvement: Description of a case and review of the literature. Clin Rheumatol. 2009 Jun;28(6):739–743.
- Groh M, Pagnoux C, Baldini C, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) (EGPA) Consensus Task Force recommendations for evaluation and management. Eur J Intern Med. 2015 Sep;26(7):545–553.
- Guillevin L, Lhote F, Gayraud M, et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore). 1996 Jan;75(1):17–28.
- McBrien CN, Menzies-Gow A. Mepolizumab for the treatment of eosinophilic granulomatosis with polyangiitis. Drugs Today (Barc). 2018 Feb;54(2):93–101.