Seven years later, she developed acute abdominal pain. Thickening of the gallbladder wall and gallstones were detected by ultrasound, leading to a diagnosis of acute cholecystitis. Her symptoms did not improve with fasting or antibiotic therapy, and she subsequently gained 10 kg weight in a month, and developed fever, dyspnea and abdominal distention. She was then admitted to our unit.
Laboratory results revealed anemia (hemoglobin 6.8 g/dL; normal range [NR] 11.3–15.2); thrombocytopenia (platelets 3.9×104/µL; NR 13.0×104–36.9×104); low albumin (2.6 g/dL; NR 3.8–5.2); renal dysfunction, requiring renal replacement therapy; and systemic inflammation (C-reactive protein [CRP] 55.6 mg/L; NR <3.0). We found slightly elevated levels of soluble interleukin (IL) 2 receptor (659 U/mL; NR 145–519), IL-6 (25 pg/mL; NR <4.0) and vascular endothelial growth factor (VEGF) (118 pg/L, NR <38.3).
Her complement levels were normal, and tests for autoantibodies, including double-stranded-DNA (dsDNA) antibodies and anti-Ro/La antibodies, were negative. Serologies for hepatitis B, hepatitis C, Epstein-Barr virus, cytomegalovirus, human immunodeficiency virus and human herpesvirus 8 (HHV-8) were negative.
A urinalysis showed hematuria and proteinuria (urinary protein to creatinine ratio 4.69 g/g·Cr) with active urine sediment, such as granular and waxy casts. Computed tomography revealed bilateral pleural effusions, massive ascites, splenomegaly and generalized lymphadenopathy (see Figure 1, below).
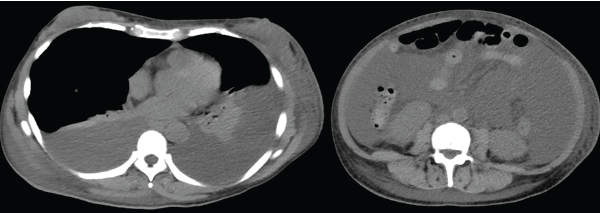
Figure 1: Computed tomography demonstrates bilateral pleural effusion and massive ascites.
The patient was initially diagnosed with SLE, based on her serositis, nephritis and pancytopenia in the presence of ANA and anti-U1 RNP antibodies. We treated her with pulse methylprednisolone (1,000 mg/day for three days), followed by prednisolone (1.0 mg/kg/day), mycophenolate mofetil (2,000 mg/day) and tacrolimus (3 mg/day), with transient hemodialysis. However, her condition gradually progressed.
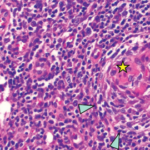
We performed a bone marrow aspiration and biopsy. Although the bone marrow aspiration was dry, the bone marrow biopsy led to the discovery of mild myelofibrosis with an increased number of slightly atypical, multi-nucleated megakaryocytes, consistent with a diagnosis of iMCD-TAFRO.
Despite treatment with intravenous tocilizumab (400 mg) and administration of a thrombopoietin receptor agonist, her symptoms did not improve. However, after administration of cyclosporine A (CsA, 3 mg/kg/day) with a target trough serum CsA level of 150–250 ng/mL, the patient finally achieved full recovery.
International, evidence-based consensus treatment guidelines for iMCD suggest tocilizumab with or without glucocorticoids as a first-line therapy for both iMCD-TAFRO & iMCD-NOS.
Discussion
Castleman disease is a heterogeneous LPD characterized by lymph node hyperplasia. Patients with Castleman disease present with solitary (unicentric) or multicentric lymphadenopathy. Around 50% of multicentric Castleman disease cases are driven by HHV-8 infection, while the etiology of the remaining half of HHV-8 negative/iMCD cases is unknown.
iMCD-TAFRO is a distinct clinical phenotype.2 A case report of iMCD-TAFRO was first described in Japan in 2010, but cases have been reported from all over the world.3 Patients with iMCD-TAFRO often have a poor prognosis; when compared with other forms of iMCD, iMCD-TAFRO is more more likely to be associated with hepatic and renal dysfunction, and a more aggressive clinical course.1
Diagnostic criteria for iMCD-TAFRO were proposed in 2019.4 A diagnosis requires all three items in the major criteria category and at least two of four items in the minor criteria category.
The major criteria include:
- Anasarca;
- Thrombocytopenia; and
- Evidence of systemic inflammation (i.e., fever of unknown etiology and elevated serum CRP).
Minor criteria include:
- Characteristic lymph node pathology;
- Reticulin myelofibrosis;
- Organomegaly; and
- Progressive kidney injury.
When diagnosing iMCD-TAFRO, the following should be excluded:
- Malignancies, including lymphoma;
- Autoimmune disorders, including SLE, Sjögren’s syndrome and ANCA-associated vasculitis;
- Infectious diseases;
- POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes);
- Liver cirrhosis; and
- Thrombotic microangiopathy.
When evaluating patients with any of these diagnoses, however, the presence of atypical features should prompt you to think of iMCD-TAFRO.
To establish a diagnosis, criteria proposed in 2015 required a lymph node biopsy, which should demonstrate the atrophy of germinal centers with enlarged endothelial cells nuclei, proliferating endothelial venules in the interfollicular zone and a few mature plasma cells.5 However, the requirement for a lymph node biopsy establish a diagnosis of iMCD-TAFRO was included as a minor, rather than a major, diagnostic criterion because the presence of anasarca and thrombocytopenia may make it difficult to perform a lymph node biopsy.
The pathogenesis of iMCD-TAFRO remains unclear.2 Some differences between iMCD-TAFRO and iMCD not otherwise specified (iMCD-NOS) have been reported. iMCD-TAFRO seems to be driven by IL-2, which causes a systemic capillary leak syndrome characterized by fluid retention, renal failure and thrombocytopenia.6 iMCD-TAFRO tends to be associated with more modest elevations of serum IL-6 and higher VEGF levels than those found in iMCD-NOS.7,8 Peritoneal levels of IL-6 and VEGF have reportedly been elevated in iMCD-TAFRO, which may contribute the development of this disease.9 Elevated serum platelet-derived growth factor level has also been reported in iMCD-TAFRO and may be associated with myelofibrosis.10
In addition, some similarities between iMCD-TAFRO and lupus have been reported. B cell and T cell activation appear to play a pathogenic role in iMCD-TAFRO which may explain the response of iMCD-TAFRO to both rituximab and cyclosporine A.2 Further, regarding epigenetic alternation, a DNA methyltransferase 3A mutation has been observed in patients with iMCD-TAFRO and SLE.11,12
Differentiating iMCD-TAFRO and lupus is critical to determine the appropriate therapeutic strategy. Approximately 10% of lupus cases meet the diagnostic criteria of iMCD-TAFRO.13 The acute and subacute manifestations of SLE and those of iMCD-TAFRO may not be distinguishable clinically, because both can be associated with systemic inflammation, effusions, cytopenias and generalized lymphadenopathy.
In a retrospective study, patients diagnosed with SLE who also met the diagnostic criteria for iMCD-TAFRO were more likely to be older, anti-dsDNA antibody negative and have higher titers of serum C-reactive protein than patients with SLE. Thrombocytopenia associated with iMCD-TAFRO was also less responsive to therapy and required longer treatment courses to achieve remission.
Therefore, physicians should consider a diagnosis of iMCD-TAFRO when evaluating a patient with SLE who is older or has a limited response to immunosuppressive therapy.
The ideal treatment for iMCD-TAFRO has not yet been established.
Clinical trials are challenging due to the rarity of patients with this disease. However, international, evidence-based consensus treatment guidelines for iMCD recommend tocilizumab with or without glucocorticoids as a first-line therapy for both iMCD-TAFRO and iMCD-NOS. In tocilizumab-resistant cases, cyclosporine A may be considered, particularly for refractory peritoneal effusion and thrombocytopenia.7
Based on retrospective research and expert opinion, the 2015 Japanese proposal of treatment strategy for iMCD-TAFRO describes glucocorticoids as first-line therapy, followed by cyclosporine A as second-line therapy—barring contraindications for cyclosporine A (e.g., renal dysfunction). If cyclosporine A is contraindicated, tocilizumab and rituximab may be considered.
Plasma exchange, cyclophosphamide (potentially as part of CHOP [cyclophosphamide, doxorubicin, vincristine and prednisolone]) and proteasome inhibitors, such as thalidomide and lenalidomide, are suggested for select patients.5
In a case series, three patients with tocilizumab-resistant iMCD-TAFRO achieved long-term remission with sirolimus, an inhibitor for mechanistic target of rapamycin (mTOR) that regulates VEGF expression and T cell activation.8
A case series reported tacrolimus may also be effective.14 Cyclosporine A and tacrolimus are both calcineurin inhibitors that regulate IL-2 transcription, leading to T cell inactivation.
In our case, however, cyclosporine A proved superior in efficacy over tacrolimus. Although further investigations are required, differences in drug delivery (i.e., penetrance into the bone marrow and effusions) and its effect on transforming growth factor β production, which increases platelet counts, may explain why cyclosporine A may be a superior therapy for iMCD-TAFRO.
Conclusion
iMCD-TAFRO may mimic SLE. An accurate differential diagnosis and consideration for an underlying autoimmune mechanism are critical to ensure appropriate disease management.
 Nobuya Abe, MD, is a clinical fellow in the Department of Rheumatology, Endocrinology and Nephrology, Graduate School of Medicine, Hokkaido University, Sapporo, Hokkaido, Japan.
Nobuya Abe, MD, is a clinical fellow in the Department of Rheumatology, Endocrinology and Nephrology, Graduate School of Medicine, Hokkaido University, Sapporo, Hokkaido, Japan.
 Yuichiro Fujieda, MD, PhD, is an assistant professor in the Department of Rheumatology, Endocrinology and Nephrology, Faculty of Medicine, Hokkaido University.
Yuichiro Fujieda, MD, PhD, is an assistant professor in the Department of Rheumatology, Endocrinology and Nephrology, Faculty of Medicine, Hokkaido University.
 Tatsuya Atsumi, MD, PhD, is a professor in the Department of Rheumatology, Endocrinology and Nephrology, Faculty of Medicine, Hokkaido University.
Tatsuya Atsumi, MD, PhD, is a professor in the Department of Rheumatology, Endocrinology and Nephrology, Faculty of Medicine, Hokkaido University.
Authors’ note: All authors contributed to the patient’s care and the writing of the report. Written consent for publication was obtained from the patient.
References
- Iwaki N, Fajgenbaum DC, Nabel CS, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am J Hematol. 2016 Feb;91(2):220–226.
- Fajgenbaum DC. Novel insights and therapeutic approaches in idiopathic multicentric Castleman disease. Blood. 2018;132(22):2323–2330.
- Takai K, Nikkuni K, Shibuya H, Hashidate H. [Thrombocytopenia with mild bone marrow fibrosis accompanied by fever, pleural effusion, ascites and hepatosplenomegaly.] [article in Japanese] Rinsho Ketsueki. 2010 May;
51(5):320–325. - Masaki Y, Kawabata H, Takai K, et al. 2019 updated diagnostic criteria and disease severity classification for TAFRO syndrome. Int J Hematol. 2020 Jan;111(1):155–158.
- Masaki Y, Kawabata H, Takai K, et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome, 2015 version. Int J Hematol. 2016 Jun;103(6):686–692.
- Konishi Y, Takahashi S, Nishi K, et al. Successful treatment of TAFRO syndrome, a variant of multicentric Castleman’s disease, with cyclosporine A: Possible pathogenetic contribution of interleukin-2. Tohoku J Exp Med. 2015 Aug;236(4):289–295.
- van Rhee F, Voorhees P, Dispenzieri A, et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood. 2018 Nov 15;132(20):2115–2124.
- Fajgenbaum DC, Langan RA, Japp AS, et al. Identifying and targeting pathogenic PI3K/AKT/mTOR signaling in IL-6-blockade-refractory idiopathic multicentric Castleman disease. J Clin Invest. 2019 Aug 13;129(10):4451–4463.
- Kubokawa I, Yachie A, Hayakawa A, et al. The first report of adolescent TAFRO syndrome, a unique clinicopathologic variant of multicentric Castleman’s disease. BMC Pediatr. 2014 Jun 2;14:139.
- Iwaki N, Gion Y, Kondo E, et al. Elevated serum interferonγ-induced protein 10 kDa is associated with TAFRO syndrome. Sci Rep. 2017 Feb 13;7:42316.
- Mavragani CP, Nezos A, Sagalovskiy I, et al. Defective regulation of L1 endogenous retroelements in primary Sjogren’s syndrome and systemic lupus erythematosus: Role of methylating enzymes. J Autoimmun. 2018 Mar;88:75–82.
- Pullabhatla V, Roberts AL, Lewis MJ, et al. De novo mutations implicate novel genes in systemic lupus erythematosus. Hum Mol Genet. 2018 Feb 1;27(3):421–429.
- Hasegawa E, Sato H, Wada Y, et al. Characterization of patients with systemic lupus erythematosus who meet the diagnostic criteria for TAFRO syndrome. Lupus. 2018 Mar;27(3):417–427.
- Shirai T, Onishi A, Waki D, et al. Successful treatment with tacrolimus in TAFRO syndrome: Two case reports and literature review. Medicine (Baltimore). 2018 Jun;97(23):e11045.