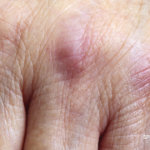
Because the rash did not improve, he was referred to a dermatologist. A skin biopsy, according to his mother, was unremarkable. The swelling gradually worsened over the next two months, and his mother noticed a new development—ulcers on his fingers (see Figure 1).
He was then referred to a rheumatologist. On examination, he had swelling of the metacarpophalangeal and proximal interphalangeal joints, ulcers at the tips of his fingers and swelling of both ankle joints. Nailfold capillaroscopy was unremarkable. His strength was 4/5 in both upper arms. The laboratory results are in Table 1. He tested negative for anti-nuclear antibodies, anti-Scl 70 antibodies and anti-topoisomerase antibodies.
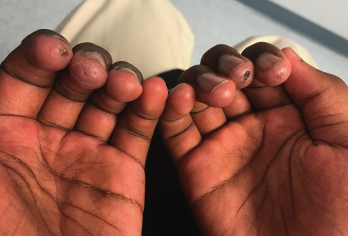
Figure 1: A 12-year-old Black male presented with digital ulcers and swelling.
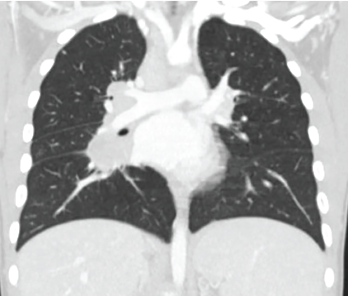
Figure 2: High-resolution computed tomography showed evidence of interstitial lung disease.
We initially thought he had systemic sclerosis. However, the lack of antibodies made diagnosis challenging. Due to lack of autoantibodies, but with muscle enzyme elevation and coexisting clinical weakness, myositis was the main diagnostic consideration.
We started treatment with 40 mg of prednisone and 10 mg of oral methotrexate (later increased to 20 mg) for myositis. Due to minimal improvement in his skin ulcers, we changed the methotrexate to a subcutaneous form at the increased dosage. However, the patient developed worsening dyspnea on exertion within a few months.
High-resolution computed tomography showed evidence of interstitial lung disease (see Figure 2). A test for anti-melanoma differentiation-associated protein 5 (MDA-5) antibody was ordered due to a high clinical suspicion of ILD. The results were positive. He was initiated on 250 mg of mycophenolate mofetil twice daily (note: 300 mg/m2, lower than the ideal 600 mg/m2 dose due to leukopenia) and 1 g/kg of intravenous immunoglobulin monthly for four doses, with significant improvement in weakness, ulcers and shortness of breath.
Table 1: Laboratory Results
| TEST | RESULT |
|---|---|
| ANA | Negative |
| Anti-Scl 70 antibody and anti-centromere antibody | Negative |
| Anti-Jo1 antibody | Negative |
| Anti-SSA 52 antibody | Negative |
| AST (aspartate aminotransferase) | 136 U/L (reference range [RR]: 15–37 U/L) |
| ALT (alanine aminotransferase) | 150 U/L (RR: 12–78 U/L) |
| Aldolase | 8.8 U/L (RR: 3.1–8.6 U/L) |
| Erythrocyte sedimentation rate (ESR) | 21 mm/hour (RR: 5–15 mm/hour) |
| Creatinine kinase | 79 mg/dL (RR: 26–308 mg/dL) |
| LD (lactate dehydrogenase) | 351 U/L (RR: 87–241 U/L) |
Discussion
Juvenile dermatomyositis is a rare autoimmune disease characterized by vasculopathy, proximal muscle weakness and pathognomonic rash. Anti-MDA-5 antibody was originally described in a subset of patients with dermatomyositis whose disease was manifested only on skin for prolonged periods of time without muscle weakness.3 The role of anti-MDA-5 antibody in dermatomyositis is not clearly known. It is thought that antibody production occurs during viral infection of skin and lung epithelium.4 Its formation is triggered by release of MDA-5 from infected cells and is believed to injure endothelial cells and other tissues.
The incidence of myositis-specific antibodies in JDM is 10%, with a prevalence of anti-MDA-5 antibody of 7.4%.5 Anti-MDA-5 antibody is associated with clinically amyopathic or hypomyopathic dermatomyositis, ischemic cutaneous ulcerations, rapidly progressive ILD and a poor prognosis. The presentation is well described in the adult population, whereas it is very rare in the pediatric population.
Rheumatologists need to be aware of anti-MDA-5 antibody-associated RP-ILD in patients with juvenile dermatomyositis, the aggressive nature of the disease and the early, intense immunosuppression needed for a favorable prognosis.