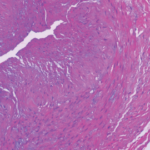
Diagnoses considered in this case included medium vessel vasculitis (e.g., polyarteritis nodosa [PAN]), segmental arterial mediolysis (SAM), fibromuscular dysplasia and collagen vascular disease.
He was started on a heparin infusion by the surgical team, and the rheumatologist recommended starting IV methylprednisolone for a possible diagnosis of medium vessel vasculitis. Exploratory laparoscopy was discussed, but on on the fifth day of his hospitalization, the patient developed chest pain, was noted to be hypotensive and had a cardiac arrest with multiple cardiac arrhythmias, including pulseless electrical activity, ventricular fibrillation and ventricular tachycardia, and unfortunately died.
The autopsy showed significant internal hemorrhage, as well as aneurysmal dilatation of the aortic arch; aneurysmal dilation, medial dissection and intramural thrombus in the major branch of the celiac artery; transmural dissection of the superior mesenteric artery and dissection of the left common iliac artery. He was found to have a gene mutation in the COL3A1 gene consistent with Ehlers-Danlos syndrome (EDS) type IV, the vascular type.
Discussion
The subtypes of EDS were defined in the 1997 Villefrache classification, based on clinical features, mode of inheritance and genetic findings. The six major types, from most to least common, are:1
- Classical;
- Hypermobility;
- Vascular;
- Kyphoscoliosis;
- Arthrochalasia; and
- Dermatosporaxis.
In 2017, an updated EDS classification was proposed by the International Consortium on Ehlers-Danlos Syndromes & Related Disorders, which now recognizes 13 subtypes.2 The Beighton hypermobility scale can help assess for classic EDS, considering hypermobility of the peripheral joints and spine.
Vascular EDS is uncommon, with inheritance almost always in an autosomal dominant manner, although the exact incidence and prevalence remain unknown. The clinical diagnosis of vascular EDS is characterized by four clinical criteria: easy bruising, thin skin with visible veins, characteristic facial features and the rupture of arteries, uterus or intestines. Unfortunately, it is often only recognized after a catastrophic complication.1,3 The diagnosis can be confirmed by culture of fibroblasts, which synthesize abnormal type III procollagen molecules, or identifying a mutation in the gene for type III procollagen, COL3A1, by sequencing. The COL3A1 gene provides instructions for making a component of type III collagen, which is found in such tissues as the skin, lungs, intestinal walls and walls of blood vessels. The resultant arterial and tissue fragility is responsible for the often catastrophic complications. Midsize arteries are most commonly involved, and arterial rupture is the most common cause of sudden death. Spontaneous arterial rupture has a peak incidence in the third or fourth decade of life, but it may occur earlier.3
Although no treatment is known or currently available, physician unfamiliarity with the disease may compromise care and lead to loss of life. Patients with vascular EDS should avoid trauma, elective surgery, arteriography and colonoscopy. Also of note, intracranial aneurysms associated with vascular EDS should be considered in the differential diagnosis of cerebral vascular disorders and stroke in early childhood, and this may have been relevant given our patient’s family history.4
Vascular EDS is an uncommon, but important consideration, when one is evaluating a patient for diseases that affect the medium-caliber blood vessels (e.g., PAN), and SAM, a non-inflammatory arterial disease. Although systemic inflammation is generally seen in PAN but not SAM, both may have a similar appearance on an angiogram, with aneurysms, dissections, stenoses and occlusions. Histopathology is the gold standard for the diagnosis of SAM, which mainly affects intra-abdominal vasculature.5 Although a challenge, identifying vasculitis is particularly vital, because other vasculopathies discussed in this article won’t respond to treatment with corticosteroids and immunosuppressive agents.
Unlike aortic dissection, which is often fatal without treatment, dissection of the SMA doesn’t have the same prognosis, possibly because there is less pressure throughout the vessel; however, this can lead to narrowing and thrombosis in the lumen of the vessel. Given the rarity, no standard therapy exists. For an acute abdomen, open surgery is needed.
Genetic testing is the gold standard for diagnosing vascular EDS.
If the patient is asymptomatic or symptomatic without evidence of bowel ischemia, SMA dissection may be treated with bowel rest and heparin, a course of treatment proposed by Ambo et al.6 Anticoagulants prevent clot and emboli formation, which help recanalize the true lumen. Antiplatelet therapy is also used to prevent arterial thrombosis. No data exist to indicate the optimal dosage or administration period. Although some say medical treatment should continue until radiographic evidence of resolution of the dissection, others suggest anticoagulation with vitamin K antagonists for one year.7,8 Unfortunately, as seen in this case, when complications, such as catastrophic vessel rupture occur, heparin administration may complicate this further.
Conclusion
It is important to think of rare collagen vascular diseases, such as vascular EDS, and non-inflammatory vessel disorders, such as SAM, when evaluating patients for a possible diagnosis of PAN. Clues to look for include external signs and family history, and you should obtain genetic testing in a timely manner. However, the window of opportunity is often small in patients with catastrophic presentation, and one may feel obligated to treat such a patient, cautiously, for a possible diagnosis of medium-vessel vasculitis, which would progress in the absence of immunosuppression. Biopsy is often not possible, and angiograms may lead to further complications. Unfortunately, this diagnosis is often established after postmortem examination.
 Catherine (Katie) Donnelly, MB, BCh, BAO, is a second-year rheumatology fellow at the University of Cincinnati Medical Center.
Catherine (Katie) Donnelly, MB, BCh, BAO, is a second-year rheumatology fellow at the University of Cincinnati Medical Center.
 Surabhi Khanna, MD, is an assistant professor of rheumatology and heads the Scleroderma Center at the University of Cincinnati Medical Center.
Surabhi Khanna, MD, is an assistant professor of rheumatology and heads the Scleroderma Center at the University of Cincinnati Medical Center.
References
- Beighton P, De Paepe A, Steinmann B, et al. Ehlers-Danlos syndromes: Revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998 Apr 28;77(1):31–37.
- Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017 Mar;175(1):8–26.
- Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000 Mar 9;342(10):673–680.
- Kato T, Hattori H, Yorifuji T, et al. Intracranial aneurysms in Ehlers-Danlos syndrome type IV in early childhood. Pediatr Neurol. 2001 Oct;25(4):336–339.
- Baker-LePain JC, Stone DH, Mattis AN, et al. Clinical diagnosis of segmental arterial mediolysis: Differentiation from vasculitis and other mimics. Arthritis Care Res (Hoboken). 2010 Nov;62(11):1655–1660.
- Ambo T, Noguchi Y, Iwasaki H, et al. An isolated dissecting aneurysm of the superior mesenteric artery: report of a case. Surg Today. 1994;24(10):933–936.
- Nagai T, Torishima R, Uchida A, et al. Spontaneous dissection of the superior mesenteric artery in four cases treated with anticoagulation therapy. Intern Med. 2004 Jun;43(6):473–478.
- Takach TJ, Madjarov JM, Holleman JH, et al. Spontaneous splanchnic dissection: Application and timing of therapeutic options. J Vasc Surg. 2009 Sep;50(3):557–563.