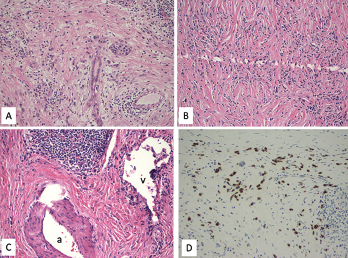
Figure 1. A. Lymphoplasmacytic infiltrate with pancreatic exocrine component atrophy. There is a cluster of remaining islet cells in the pancreatic duct. H&E magnification x200; B. Dense fibrosis with storiform pattern. H&E magnification x200; C. Lymphoplasmacytic infiltrate of a vein (phlebitis) without obliteration of the lumen. H&E magnification x200; v—vein, a—artery; D. Immunostain for IgG4 highlights increased IgG4-positive plasma cells, up to 80 cells/HPF. Immunostain, magnification x200.
Sarcoidosis and IgG4-related disease (IgG4-RD) are both immune-mediated, often multi-organ, diseases of uncertain etiology capable of presenting with diverse clinical manifestations. Many clinical features are common to both conditions, including hypergammaglobulinemia, the ability to form inflammatory masses and involvement of the lymph nodes, lacrimal glands, salivary glands, meninges and lungs. Although imaging modalities, such as positron emission tomography/computed tomography (PET/CT), and serologic evaluation may suggest one disease over the other, these diagnoses remain dependent on tissue sampling and histopathologic examination. Histopathologic evaluation is also essential to rule out other mimicking conditions, such as lymphoma.
Here, we present the case of a patient previously diagnosed with IgG4-RD, after presenting with a pancreatic mass, who was subsequently diagnosed with sarcoidosis based on a biopsy of a mediastinal lymph node.