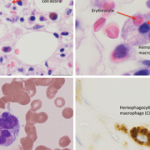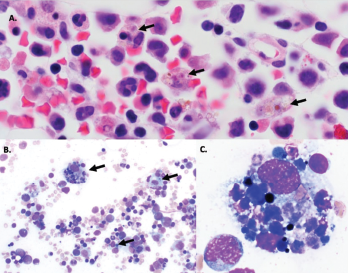
Figure 1A–C: Arrows denote hemophagocytic histiocytes.
Figure A: Hematoxylin and eosin stained bone marrow biopsy (1,000X).
Figure B: May-Gurenwald-Giemsa bone marrow aspirate (100X).
Figure C: May-Gurenwald-Giemsa bone marrow aspirate (1,000X).
COG4 mutations have previously been reported in two patients with autosomal recessive COG4-CDG (CDG-IIj), who were described as having a similar set of clinical symptoms of hypotonia, seizures, coagulopathy and liver dysfunction, as well as recurrent infections.
Over the next three months, the patient experienced two more episodes of fever, liver dysfunction, coagulopathy and shock physiology. The second episode was preceded by a similar set of symptoms of fever, vomiting and diarrhea, and was successfully treated with FFP.