Clinical Features
Although the clinical phenotype of CVID may be heterogeneous, one key characteristic of this disease is an impaired ability to produce antibodies. This disturbance manifests in decreased levels of immunoglobulin (Ig) G, IgA, and, sometimes, IgM isotypes and in an impaired or even absent antibody response upon antigenic challenge. This results in a failure to respond to vaccinations and an increased susceptibility to infections. Infections are usually localized in the respiratory tract and a majority of patients have recurrent bronchitis, sinusitis, or otitis (which is more common in childhood-onset CVID), and have had one or more episodes of pneumonia.
Typical pathogens affecting patients with CVID are encapsulated bacteria like Haemophilus influenzae, Streptococcus pneumoniae, or Moraxella catharralis. A significant number of CVID patients also suffer from gastrointestinal tract infections caused by Giardia lamblia, Campylobacter, or Salmonella species. Pulmonary symptoms of CVID may be accompanied by an obstructive lung phenotype (e.g., chronic bronchitis) and asthma. The lung represents the Achilles’ heel of the CVID patient and chronic lung damage (e.g., bronchiectasis, lung fibrosis, and emphysema) is one of the most common causes of morbidity and mortality in the CVID cohort.
The respiratory system of CVID patients may also be affected by granulomatous inflammation, characterized by sarcoid-like lesions in lymph nodes and other histology specimens. This accompanying disease is found in approximately 10% to 22% of CVID patients. In addition to the lung, virtually any other organ system can be affected, but the involvement of lung and liver in particular worsens the patient’s prognosis and is often difficult to treat. High-resolution computer tomography is the method of choice to evaluate and monitor the status and progression of lung-related pathology in CVID patients.
Gastrointestinal diseases are frequent in CVID patients and manifest as chronic diarrhea, malabsorption, and weight loss. A significant number of patients suffer from inflammatory bowel disease.
The increased risk of developing autoimmune diseases underscores the sometimes paradoxical immune disregulation observed in CVID patients. The most common autoimmune manifestations in CVID are idiopathic thrombocytopenic purpura (ITP) and autoimmune hemolytic anemia (AIHA). ITP or AIHA may occur years before a patient presents with an immunodeficiency.4 A subgroup of CVID patients clinically presents with a combination of ITP/AIHA, splenomegaly, and granuloma formation.5 Other observed autoimmune diseases include pernicious anemia, autoimmune thyroiditis, rheumatoid arthritis, and vitiligo.
CVID patients have an increased risk of developing malignancies, especially lymphoma and gastric cancer, and patients should be screened regularly for these potential complications.3
Immunological Features
During the past five decades, numerous studies have identified a plethora of immunological abnormalities in patients with CVID. CVID pathogenesis has been attributed to defects in T cells and their subsets, antigen presenting cells, and B cells. Despite convincing evidence that alterations of T cell function and subset distribution are associated with the CVID phenotype, recent research points to the impaired terminal differentiation of B lymphocytes as the hallmark of the disease.5-7
Several groups have now demonstrated that the formation of memory B cells is severely impaired in the majority of CVID patients and leads to a concomitant depletion of long-lived plasma cells and a drop of serum Ig levels. The altered distribution of B cell subsets observed in CVID patients is now applied in disease classification.5-7
Diagnosis
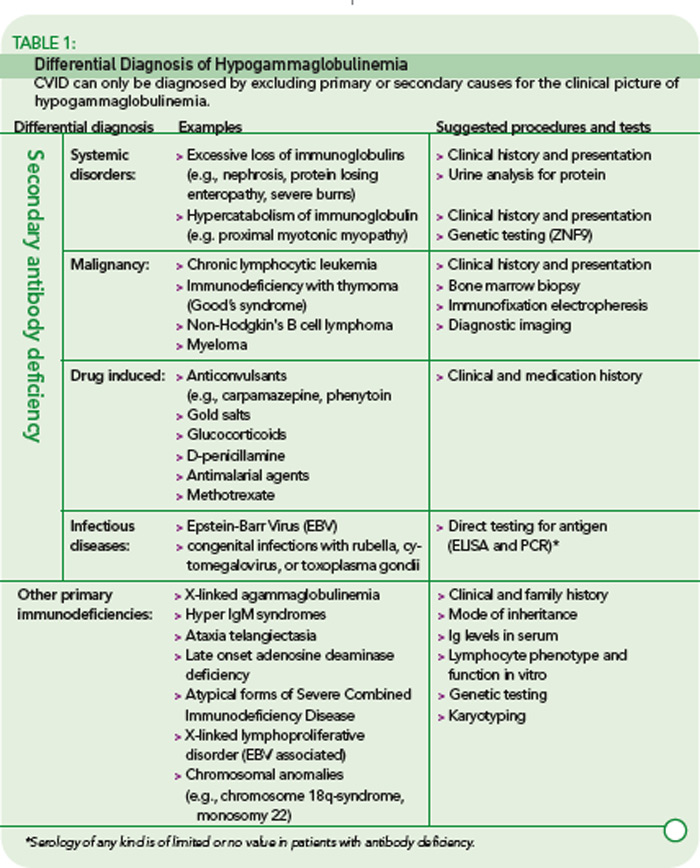
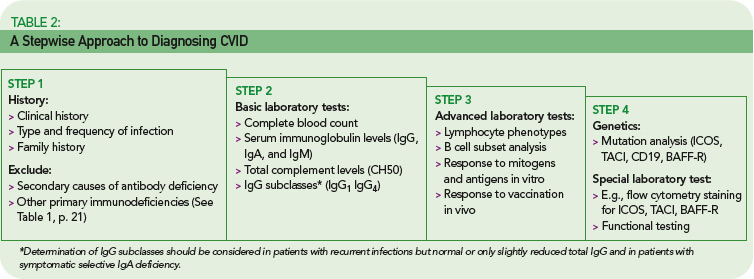
The rare incidence and high clinical variability of CVID can present a diagnostic challenge. Because there is no specific test for diagnosing CVID, diagnosis is made by excluding similar conditions. First, the clinically more frequent secondary causes for hypogammaglobulinemia must be ruled out (See Table 1, below). Then other primary immunodeficiency disorders have to be excluded, and suspected CVID should be validated, as outlined in Table 2 (p. 22).
The first step is the assessment of the clinical and family history, including a detailed description of the type, duration, and frequency of infections. In addition to recurrence of infections, unusual severity and development of complications (e.g., empyema, bronchiectasis) can indicate an underlying immune defect. Identifying underlying pathogens is very helpful in evaluating any immunodeficient patient, with infections by encapsulated bacteria being the most common type in CVID. Whenever possible, use direct detection methods for pathogens (e.g., microbial culture, antigen-ELISA, PCR) to evaluate the cause of infection, because serology is futile in patients with an antibody deficiency.
A systematic assessment of the patient’s immunological status is the next step. This includes basic laboratory analyses such as a complete blood count, serum immunoglobulin levels (IgG, IgA, and IgM), and total complement levels (CH50). The determination of IgG subclasses (IgG1 to IgG4) is especially useful in patients who have only slightly decreased or low-normal IgG serum levels but suffer from recurrent infections.
Next, perform a quantitative flow cytometric analysis of lymphocyte phenotypes. This analysis should include T cell subsets (CD4+, CD8+, and memory) as well as total (CD19+) B cell numbers and B cell subsets. The numbers of class-switched memory B cells (CD27+/IgD–), non-switched memory B cells (CD27+/IgD+), and other peripheral B cell subsets provide useful information. While total numbers of B cells in CVID are usually normal or only slightly reduced in about 90% of patients, class-switched memory B cells are reduced in up to 75% of CVID patients.6,7
Of particular importance is the assessment of specific antibody responses to different antigens (protein and polysaccharide antigens) upon vaccination.8 The results of these tests are helpful in determining whether a patient requires immunoglobulin replacement therapy, especially when a patient is hypogammaglobulinemic but has not yet had recurrent infections.
Genetic testing and specific in vitro tests (e.g., flow cytometry studies of surface markers and specialized functional testing) are available in immunodeficiency centers and specialized laboratories. Although genetic tests for CVID are only accessible to a small subgroup of patients so far, the recent discovery of the first single gene defects permits a definite diagnosis of CVID for the first time.
New Genetic Insights in the Pathogenesis of CVID
It has long been known that CVID also has a genetic component. While most cases of this disease occur sporadically, in 10% to 20% of CVID cases, at least one additional family member either suffers from CVID or selective IgA deficiency.9 With a ratio of about 4 to 1, autosomal dominant inheritance is more common than recessive inheritance in CVID families. Genetic linkage studies have revealed that the genetic defect involved in this disease cannot be reduced to a single gene locus.
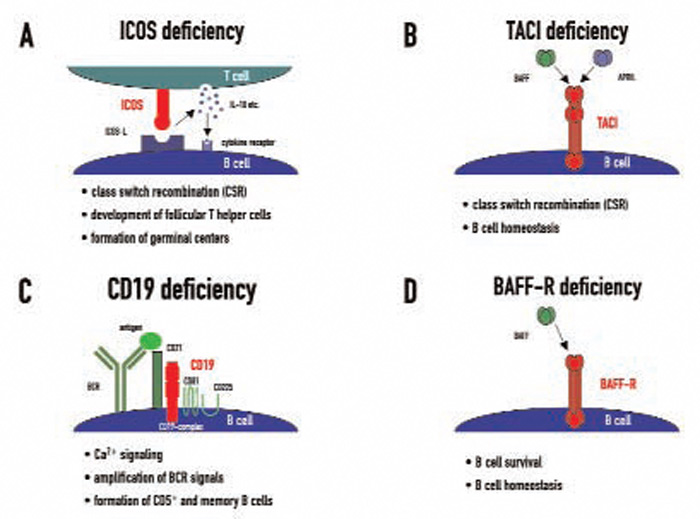
Candidate loci for CVID have now been demonstrated at the HLA region on chromosome 6, chromosome 4q, and chromosome 16q. This genetic heterogeneity—which probably mirrors the variable clinical presentation of this disease—is further increased by the recent discovery of four candidate genes which were found to be mutated in CVID independently of the results of previous linkage and association studies. These genes—ICOS, TACI, CD19, and BAFFR—encode for cell surface receptors on lymphocytes that play crucial roles either in peripheral B cell development or in B cell function.9
ICOS (inducible costimulator on activated T cells) was the first gene defect described in patients with CVID and has so far been described in nine individuals from four families, all of whom inherited the same mutation from a common founder.10 ICOS is expressed on activated T cells and is of critical importance for correct T cell–B cell cooperation during the adaptive immune response. It belongs to the family of T cell co-stimulatory molecules and interacts with ICOS-L on B cells, leading to the release of IL10, IL-17, and several other cytokines from T lymphocytes (See Figure 1A, p. 22). These cellular signals enable B cells to undergo class switch recombination.
In addition to the effects on class switch recombination, ICOS-L/ICOS interactions seem to be crucial for the development of a highly specialized CD4+ T cell subpopulation called follicular T helper cells, which direct the formation of germinal centers in peripheral lymphoid organs.11 ICOS expression loss causes functional germinal center development to fail and therefore leads to impaired terminal B cell differentiation and hypogammaglobulinemia. Childhood onset of the disease was observed in one ICOS-deficient family. All but one adult ICOS-deficient patient had severe peripheral B cell lymphopenia and almost no memory B cells.12
Transmembrane activator and calcium-modulating cyclophilin ligand interactor (TACI) deficiency was the second molecular genetic defect found in patients with CVID.13,14 TACI, which is encoded by the gene TNFRSF13b, is a member of the tumor necrosis factor (TNF) receptor superfamily. TACI interacts with the ligands BAFF/Blys (B cell activating factor, B lymphocyte stimulator) and/or APRIL (A proliferation inducing ligand)—both members of the TNF ligand superfamily. Together with its closest relatives—BAFF-R and BCMA (B cell maturation antigen)—TACI forms a complex signaling network. TACI directs immunoglobulin class switch recombination and negatively regulates B cell homeostasis (See Figure 1B, p. 22). It has been shown that TACI-deficient mouse models develop lymphoproliferation, autoimmunity, and immunodeficiency, a triad reminiscent of the clinical phenotype observed in some CVID patients.
In about 2% to 3% of CVID patients, both TACI alleles are mutated, causing receptor function loss and ultimately resulting in an antibody deficiency. Simple heterozygous TACI mutations are found in up to 8% of CVID patients. These heterozygous changes are believed to act as disease modifiers rather than causing the disease because they are also observed in the normal population, albeit at lower frequencies.15 The clinical spectrum in TACI deficiency is diverse and there is no specific B cell or immunological phenotype. TACI-deficient individuals may be prone to develop lymphoproliferative and autoimmune complications.16 To unravel the genetic complexity and mechanism of particular TACI mutations and to correlate this to the clinical and immunological presentation of TACI-deficient CVID patients is a main research focus for my colleagues and I.
CD19 deficiency is a very rare cause of CVID and, up to now, has only been described in four patients worldwide.17,18 The CD19 molecule is expressed solely on B cells and is therefore one of the most widely used surface markers to identify the B cell lineage in flow cytometry. CD19 forms a receptor complex with CD21 (CR2), CD81, and CD225 (Leu13), which is referred to as the CD19-complex. This signaling complex fine-tunes and amplifies B cell receptor (BCR) signals after antigen binding (See Figure 1C, below). CD19-deficient patients have decreased or even no expression of CD19 on the surface of their B cells and an impaired antigen-dependent Ca2+ signaling, resulting in diminished B cell responses upon BCR triggering. In these patients, the formation of memory B cells and CD5+ B cells is also reduced, consequently leading to hypogammaglobulinemia and absent vaccine responses in vivo.
My colleagues and I were able to identify two cases of homozygous BAFFR deficiency in one single kindred.19 BAFF-R is closely related to TACI and BCMA and interacts exclusively with BAFF. The BAFF-BAFFR interaction mediates signals important for B cell survival and regulates peripheral B cell homeostasis (See Figure 1D, below). BAFF-R deficiency presents with a distinct immunological phenotype which had been inferred by BAFF-R-deficient mouse models: peripheral B cell numbers are low, transitional B cell numbers are proportionally increased, and IgA production is intact.
Therapeutic Management of CVID
The primary objective of CVID therapy is reducing the frequency and duration of recurrent and chronic infections, especially sinusitis, bronchitis, otitis, pneumonia, and gastrointestinal infections and their sequelae. Immunoglobulin replacement therapy is the treatment of choice and forms the foundation of therapy.
The standard dosing recommendation for intravenous immunoglobulin (IVIG) is 400 mg per kilogram body weight every three to four weeks. Immunoglobulins may also be administered subcutaneously in weekly intervals. An IgG trough level (the IgG level before the next infusion) of at least 5 g/L should be attained. The individual patient’s clinical history and outcome will determine the amount of IVIG required; some patients (e.g., those with bronchiectasis, diarrhea, or IgG hypercatabolism) may require higher IVIG doses to reach the mandatory trough level. To maximize treatment success, the dose, frequency, way of administration, and product type should be adapted to each individual needs.
Antimicrobial therapy is the other main component of CVID therapy, because immunoglobulin replacement alone may not adequately prevent or treat local and/or persistent infections. Consider prolonged and intensified therapeutic regimens (e.g., intravenous antibiotics for patients with known bronchiectasis).
In addition, the accompanying diseases and sequelae of CVID require adequate treatment. Corticosteroids and cyclosporine A are effective for granulomatous manifestations and autoimmune diseases, although long-term treatment efficacy may be limited due to side effects.
From recent case reports, novel monoclonal anti-B-cell antibodies are promising new agents to combat autoimmune and granulomatous complications in CVID. However their effectiveness needs to be assessed in systematic double-blind, randomized clinical trials.
Conclusion
CVID comprises a genetically and clinically heterogeneous group of diseases in which diagnosis is often delayed or even missed. In the majority of cases, CVID remains a clinical diagnosis of exclusion. However, the identification of the first genetic defects associated with a CVID phenotype mark substantial progress for clinical immunology. These findings not only allow a definite diagnosis in a small subgroup of patients, but also serve as models for understanding the pathogenesis of CVID as well as the function of the normal immune system. The discovery of additional genes associated with CVID and the mechanisms by which the mutation promotes CVID will be essential for the future diagnosis and treatment of this disease. Genetic testing may also reduce the time delay between onset of initial symptoms and diagnosis, improving patient outcomes.
Drs. Myrtek and Salzer are researchers in the division of rheumatology and clinical immunology at the Medical School of the University Hospital Freiburg in Hugstetterstr, Germany.
References
- Janeway CA, Apt L, Gitlin D. Agammaglobulinemia. Trans Assoc Am Physicians. 1953;66:200-202.
- International Union of Immunological Societies. Primary immunodeficiency diseases. Report of an IUIS Scientific Committee. Clin Exp Immunol. 1999;118:1-28.
- Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. 1999;92:34-48.
- Wang J, Cunningham-Rundles C. Treatment and outcome of autoimmune hematologic disease in common variable immunodeficiency (CVID). J Autoimmun. 2005;25:57-62.
- Wehr C, Kivioja T, Schmitt C, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. 2007; doi:10.1182/blood-2007-06-091744
- Piqueras B, Lavenu-Bombled C, Galicier L, et al. Common variable immunodeficiency patient classification based on impaired B cell memory differentiation correlates with clinical aspects. J Clin Immunol. 2003;23:385-400.
- Warnatz K, Denz A, Drager R, et al. Severe deficiency of switched memory B cells (CD27(+)IgM(-)IgD(-)) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood. 2002;99:1544-1551.
- Goldacker S, Draeger R, Warnatz K, et al. Active vaccination in patients with common variable immunodeficiency (CVID). Clin Immunol. 2007;124:294-303.
- Schäffer AA, Salzer U, Hammarström L, Grimbacher B. Deconstructing common variable immunodeficiency by genetic analysis. Curr Opin Genet Dev. 2007;17:201-212.
- Grimbacher B, Hutloff A, Schlesier M, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol. 2003;4:261-268.
- Bossaller L, Burger J, Draeger R, et al. ICOS deficiency is associated with a severe reduction of CXCR5+CD4 germinal center Th cells. J Immunol. 2006;177:4927-4932.
- Warnatz K, Bossaller L, Salzer U, et al. Human ICOS deficiency abrogates the germinal center reaction and provides a monogenic model for common variable immunodeficiency. Blood. 2006;107:3045-3052.
- Salzer U, Chapel HM, Webster AD, et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet. 2005;37:820-828.
- Castigli E, Wilson SA, Garibyan L, et al. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet. 2005;37:829-834.
- Pan-Hammarström Q, Salzer U, Du L, et al. Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat Genet. 2007;39:429-430.
- Zhang L, Radigan L, Salzer U, et al. Transmembrane activator and calcium-modulating cyclophilin ligand interactor mutations in common variable immunodeficiency: clinical and immunologic outcomes in heterozygotes. J Allergy Clin Immunol. 2007;120:1178-1185.
- van Zelm MC, Reisli I, van der Burg M, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med. 2006; 354:1901-1912.
- Kanegane H, Agematsu K, Futatani T, et al. Novel mutations in a Japanese patient with CD19 deficiency. Genes Immun. 2007; doi: 10.1038/sj.gene.6364431
- Warnatz K, Salzer U, Gutenberger S, et al. Finally found: human BAFF-R deficiency causes CVID. XIth Meeting of the European Society for Immunodeficiencies. 2004;21-24.