Correct!
B) Pigmented purpuric dermatosis (PPD)
PPD is a relatively common dermatologic condition. Distinguishing vasculitis and PPD is most relevant, as the two can be easily clinically confused. Traditionally, there are five subtypes of PPD (See Table 1). Considerable clinical and histopathologic overlap may exist between subtypes. All subtypes of PPD share common features, including:
- They are benign disorders.
- There are no associated systemic symptoms.
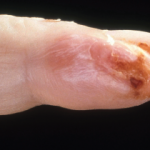
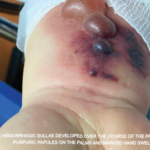
- The characteristic appearance includes petechiae and/or violaceous or copper-brown macules and papules.
- Lesions tend to occur in dependent areas, such as the lower extremities and buttocks, but can be seen anywhere on the skin surface.
- Though the etiology is not well understood, the skin changes are thought to represent perivascular inflammation and deposition of hemosiderin.1 Vasculitis, with vessel wall damage and fibrin deposition, is not a feature.
The most common subtype of PPD is Schamberg’s purpura. This eruption is characterized by copper-brown macules covered with pinpoint-sized petechiae, often described as resembling cayenne pepper. This subtype is most commonly seen on the lower legs of older men, although cases in children have been reported.2 Pigmented purpuric lichenoid dermatitis (Gougerot-Blum syndrome) is a variant that clinically resembles Schamberg’s purpura, but lesions tend to be raised and more deeply violaceous, which is likely due to the presence of a lichenoid band seen histologically. Lichen aureus is another subtype of PPD that demonstrates a lichenoid band histologically. This variant manifests with the sudden onset of golden-brown macules and papules that do not blanch with pressure. Segmental subtypes of this variant have been described.3 A variant known as Ducas and Kapetanakis pigmented purpura is characterized by eczematous scaly patches with petechiae, and is the most likely subtype to be associated with pruritus. Purpura annularis telangiectodes (Majocchi’s disease) is perhaps the most clinically distinctive of the five subtypes, and presents with annular blue–red scaly patches with petechiae. These patches can extend peripherally and become atrophic at the center. This is the only subtype that is thought to occur more frequently in adolescent females.4 Several other rare types of PPD have been described, including granulomatous, linear, familial, and transitory, though these types are rare.5
The precise pathogenesis of PPD is unknown. It has been speculated that cell-mediated immunity and Langerhan cells play an important role in inciting inflammation.5,6 Histologically, there is a lymphocytic infiltrate surrounding superficial vessels, along with hemosiderin deposition within macrophages and endothelial swelling with erythrocyte extravasation. There is no vessel wall destruction or fibrin deposition.5,7 Tissue specimens can demonstrate a variable amount of spongiosis or a lichenoid infiltrate depending on the subtype.