The rheumatologist is frequently called upon to evaluate patients with complaints of myalgia, muscle cramping, and fatigue. Because these symptoms may be nonspecific and lack any clear temporal or anatomic pattern, their workup may entail costly and uninformative tests. When similar symptoms emerge during or following physical exertion, a metabolic myopathy should be suspected. Recurrent myoglobinuria, exercise intolerance, and mild fixed proximal muscle weakness are also frequently encountered in metabolic myopathies. Although inflammatory myopathies may present in a similar fashion, such a pattern should prompt a thorough evaluation for an underlying metabolic myopathy. This review will discuss an approach to the diagnosis and treatment of several of the more common metabolic myopathies.
Metabolic Myopathies
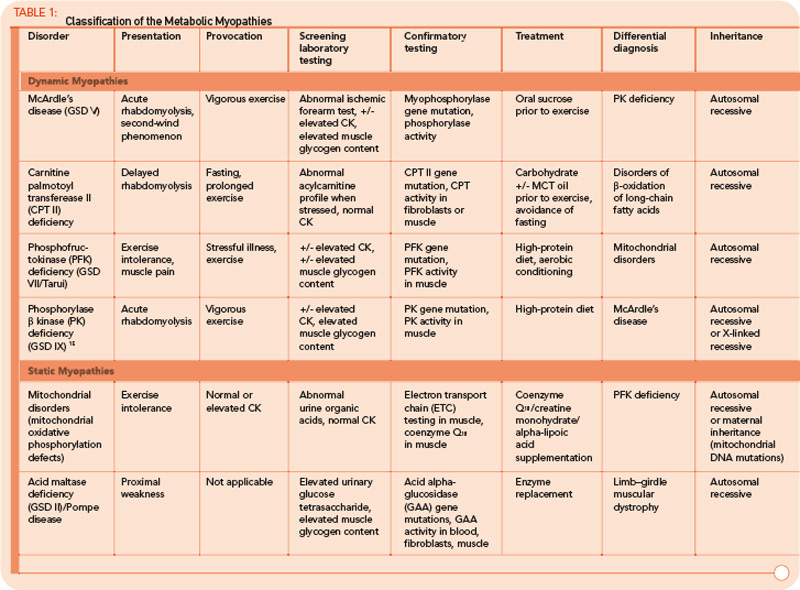
The metabolic myopathies are a heterogeneous group of disorders that share the common feature of inadequate production of cellular energy in the muscle. (See Table 1, p. 16, for a summary of metabolic myopathy classification.) They are often categorized into hereditary (primary) disorders, the focus of this review, and acquired (secondary) disorders. A further clinical distinction can be made between those disorders associated with primarily dynamic features (e.g., transient, exercise-induced fatigue, cramping, and rhabdomyolysis) and those disorders associated with primarily static features (fixed weakness).
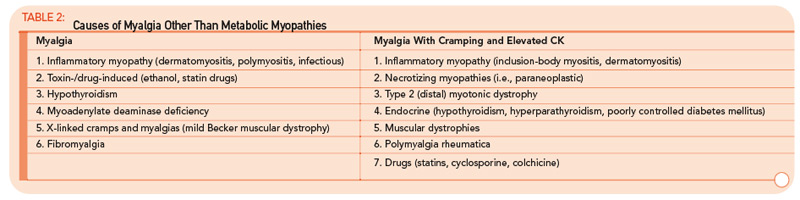
The variable and often transient nature of these symptoms can present a challenging diagnostic dilemma. Depending on the underlying defect, some patients may present in infancy or early childhood, whereas others may not present until well into adulthood. Some disorders may be strictly limited to muscle pathology, whereas others, particularly those involving mitochondrial dysfunction, may demonstrate multisystem involvement. Furthermore, a number of acquired and inherited conditions may mimic metabolic myopathies and must be properly diagnosed both to allow appropriate treatment and counseling and in order to avoid unnecessary and potentially harmful treatments (see Table 2, p. 23). Recently, drug therapy for hypercholesterolemia with statins has emerged as a cause for iatrogenic myalgia.2 The mechanism of muscle injury is not known, but there appears to be a dose-related effect, with higher statin doses more frequently leading to myopathy and rhabdomyolysis. A recent study has also associated an increased risk for statin-induced myopathy in patients harboring a common single-nucleotide polymorphism in the SLCO1B1 gene on chromosome 12, which is related to hepatic uptake of statin drugs.3 The incidence of alleles for rare metabolic myopathies, either in homozygotes or heterozygotes, was increased in a large group evaluated for statin-induced myopathy, and muscle coenzyme Q10 deficiency was highly prevalent in that group.4 Whether the coenzyme Q10 deficiency responds to supplementation remains to be established, but the biochemical abnormality could represent an important lead regarding the pathogenesis of statin-induced myopathy.
Other myopathic conditions may mimic metabolic myopathies, in particular the limb–girdle muscular dystrophies or, more commonly, chronic inflammatory myopathies such as polymyositis. For a more detailed discussion, Wortmann and DiMauro provide an excellent review of the subject.1 In general, other systemic features such as rash, malaise, interstitial fibrosis, carditis, and gastrointestinal dysfunction should suggest the presence of an acquired inflammatory myopathy, but the distinction can be difficult.
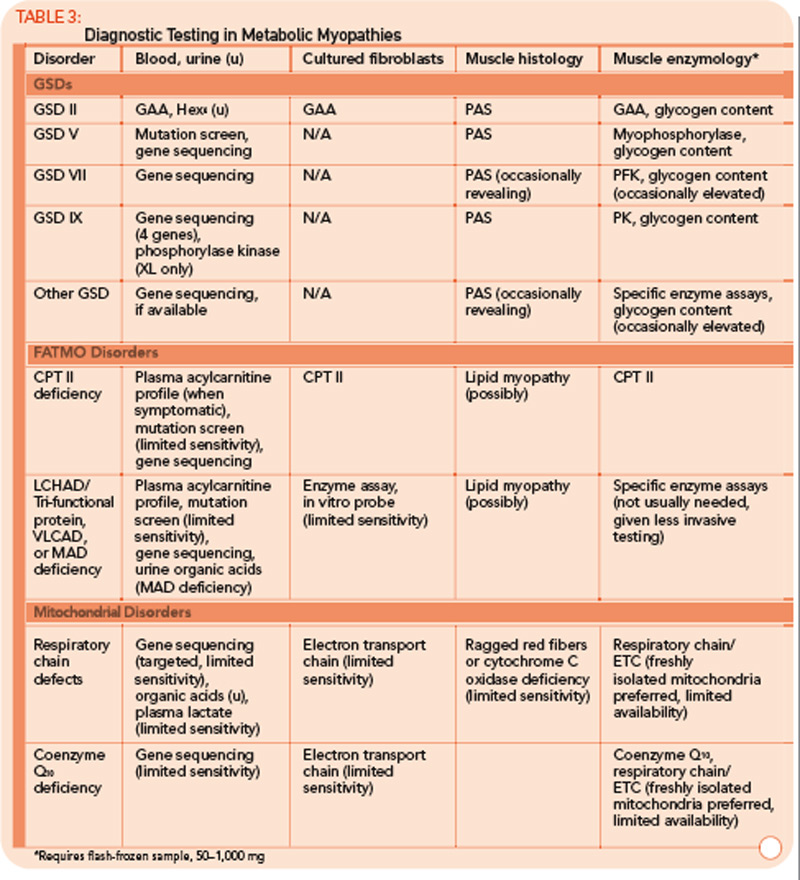
Many tools are available to the clinician to aid in the diagnostic evaluation of metabolic myopathies. One of the most cost-effective and least invasive tools remains the clinical history and physical examination, which can provide invaluable information that allows the clinician to narrow or broaden further diagnostic testing as appropriate and potentially avoid unnecessary invasive testing. A thorough history and examination will often help differentiate a dynamic myopathy characterized by transient, severe symptoms from a static myopathy characterized by fixed weakness. Because the primary metabolic myopathies are hereditary, a thorough, targeted family history may provide valuable information. Routine tests including serum electrolytes, glucose, liver transaminases, creatine kinase (CK), lactate, ammonia, and urinalysis may be useful. In some cases, the forearm exercise test, electromyography (EMG), and routine muscle biopsy may provide valuable information (see Table 1, p. 16, and Table 3, p. 23). Diagnostic tests such as urine organic acids, plasma amino acids, and plasma acylcarnitine profile may provide more specific results, allowing the clinician to approach more costly confirmatory testing, including enzyme and DNA analysis on leukocytes, fibroblasts, and the liver, in a rational, cost-effective manner (see Table 3, p. 23).
As a group, the metabolic myopathies are often broadly divided into three categories based on the underlying metabolic defect: 1) muscle glycogenoses, 2) disorders of FAO, and 3) mitochondrial myopathies. The following discussion will focus on the most common primary metabolic myopathies associated with primarily dynamic symptoms characterized by exercise-induced myalgia, early fatigue, cramping, and myoglobinuria.
Disorders of Glycogen Metabolism (Muscle Glycogenoses)
The classification of GSDs is based on the associated enzyme defect and clinical presentation.5 Two GSDs do not involve skeletal muscle: GSD type I (von Gierke disease) and GSD type VI (Hers disease). Some GSDs produce primarily static symptoms of fixed, proximal weakness, including GSD type II (Pompe disease), GSD type III (Cori-Forbes disease), and GSD type IV (Andersen disease). The latter two disorders are typified by liver involvement and will not be further discussed. Late-onset Pompe disease causes progressive, proximal muscle weakness that may progress to respiratory failure, and it falls into the static myopathy category.
Phosphofructokinase deficiency (GSD type VII; PFK deficiency or Tarui disease) might be underdiagnosed because it presents with nonspecific exercise intolerance and myalgias. Until recently, Tarui disease could only be diagnosed by functional enzyme testing requiring muscle biopsy. The diagnostic evaluation has been simplified with the recent availability of gene testing. The remaining disorders are quite rare, with the exception of McArdle’s disease (GSD type V), and typically produce dynamic symptoms. Given the relatively common occurrence and characteristic clinical presentation, we will discuss McArdle’s disease in more detail here.
Case 1
A 22-year-old male presented to the neuromuscular clinic with exertional myalgia and recurrent myoglobinuria. When the patient was a young child, his first symptoms were painful, transient contractures of his forearm finger flexors noted when climbing trees. Attacks became more frequent with age, and he first noticed dark urine associated with muscle cramping as a high school student. He did not report symptomatic improvement following short periods of rest. Physical examination was normal in clinic. There was no weakness, and no fasciculations or fixed contractures. His medical history and family history were noncontributory. Laboratory examination revealed an elevated CK (8,404 U/L; normal, 30–220 U/L), mild elevations of aspartate aminotransferase (75 U/L; normal, 15–41 U/L), and alanine aminotransferase (82 U/L; normal, 17–63 U/L). Urinalysis was negative for myoglobin. Routine muscle histology was unremarkable, but histochemical stains were not performed. EMG demonstrated mild myopathic changes. Molecular genetic testing revealed homozygosity for the common p.R49X mutation in the myophosphorylase (PYGM) gene.
The patient was diagnosed with McArdle’s disease, instructed to avoid isometric exercises, and encouraged to engage in regular aerobic exercise with a brief warm-up period. He was also instructed to consume a high-protein, low-carbohydrate diet with frequent small meals throughout the day, to ingest the equivalent of 40 grams of sucrose 30 minutes before vigorous exercise and to avoid statin medications.
McArdle’s Disease
GSD type V (McArdle’s disease or myophosphorylase deficiency) is caused by homozygous mutations in the PYGM gene, which encodes the skeletal muscle isoform of glycogen phosphorylase, also known as myophosphorylase, an essential enzyme for glycogenolysis. Because the heart and liver isoforms are not affected, the disease exclusively affects skeletal muscle.5 The typical age of onset is in late childhood. Patients develop symptoms during brief, isometric exercise such as weightlifting or during prolonged moderate intensity exercise such as jogging or swimming. Improvement in symptoms after a brief rest, the so-called “second-wind” phenomenon, is frequently reported. Approximately 50% of patients develop fixed weakness and muscle atrophy with aging.6
Serum CK levels are usually elevated at baseline in McArdle’s disease. EMG is typically normal. The forearm exercise test will demonstrate a normal rise in ammonia levels without a rise in lactate levels. A similar pattern is seen in the other muscle GSDs with the exception of Pompe disease, brancher enzyme deficiency (GSD type IV), and phosphorylase b kinase deficiency (GSD type IX). Muscle biopsy will demonstrate absent or severely reduced myophosphorylase staining on histochemical analysis.6 Enzyme analysis of flash-frozen muscle can confirm the diagnosis by demonstrating absent or reduced myophosphorylase activity. Current molecular genetic testing on blood can detect mutations that account for about 90% of cases, avoiding the need for biopsy.5
In the absence of well-established treatment of McArdle’s disease, several therapeutic interventions have been proposed. Supplementation with creatine monohydrate, branched-chain amino acids, or pyridoxine (vitamin B6) have been reported to be beneficial, although results have been variable.7 A diet rich in complex carbohydrates combined with regular aerobic exercise is advocated by many investigators. Recently, it has been clearly shown that consumption of approximately 40 grams of sucrose or fructose shortly before vigorous physical exercise dramatically reduces symptoms.8,9 Intense isometric exercise limited to isolated muscle groups should be avoided.8,9
FAO Disorders
FAO (b-oxidation of fatty acids) is the major source of energy during periods of sustained, low-intensity exercise or prolonged fasting. In infants, these disorders typically present during periods of illness or poor oral intake. In children and adults, exercise intolerance and myoglobinuria are the most common presenting features. The major disorders of lipid metabolism that present with isolated myopathy include: 1) carnitine palmitoyltransferase II (CPT II) deficiency, 2) long-chain acyl-CoA dehydrogenase (LCHAD), 3) trifunctional protein including LCHAD, and 4) very long-chain acyl-CoA dehydrogenase (VLCAD) defects.10 Of these, CPT II deficiency is the most prevalent and is the most common overall cause of hereditary recurrent myoglobinuria.
Case 2
A 69-year-old female presented to the metabolic clinic following admission through the emergency room after a day of moving boxes on a cold December day. Symptoms included muscle pain accompanied by very dark colored urine. Physical examination was noncontributory. Initial laboratory evaluation revealed serum CK elevated to 32,000, elevated hepatic transaminases, and myoglobinuria. Pertinent medical history included initiation of atorvastatin therapy one week earlier for hypercholesterolemia, absence of previous episodes of muscle pain, and hypothyroidism treated with levothyroxine. Subsequently, atorvastatin was discontinued. Family history was negative for exercise intolerance or other neuromuscular symptoms. Laboratory testing obtained at the initial clinic visit revealed normal serum CK, plasma lactate, plasma carnitine, and plasma acylcarnitine profile. CPT II gene sequencing revealed a single inactivating mutation, which implied either carrier or affected status. Enzyme testing of cultured fibroblasts demonstrated a deficiency of carnitine palmitoyltransferase II, and the patient was advised to avoid prolonged fasting and to ingest beverages containing carbohydrates prior to and during any significant exercise. She experienced two episodes of rhabdomyolysis requiring hospitalization over the next three years. In contrast with the earlier normal acylcarnitine profile, subsequent acylcarnitine profiles revealed elevated long-chain acylcarnitines in a pattern consistent with CPT II deficiency (C16, C18:1, and C18:2).
CPT II Deficiency
CPT II is a protein located on the inner surface of the inner mitochondrial membrane and serves a critical role in the conversion of long-chain acylcarnitines back into long-chain acyl-CoA species. Long-chain acyl-CoA species subsequently undergo b-oxidation, which provides acetyl-CoA to the tricarboxylic acid cycle, driving synthesis of ATP via the electron transport chain.
CPT II deficiency can be classified into three different presentations: 1) a lethal neonatal form involving multi-organ failure, 2) a severe infantile hepatocardiomuscular form, and 3) a milder, purely myopathic form. The severity of disease appears to be related to the type of mutation. Missense mutations that allow production of some partially functional enzyme activity are typically found in the milder myopathic form, while protein truncating mutations produce the more severe neonatal and infantile phenotypes.10
In contrast to the muscle glycogenoses, the myopathic form of CPT II deficiency often does not produce cramps, and muscle weakness should not be present between attacks. Serum CK levels are usually normal, as are EMG and muscle pathology. Patients may complain of muscle weakness and myalgia triggered by exercise, infection, or fasting. Other triggers include cold exposure and general anesthesia. Age of onset is typically in childhood but can vary widely. Diagnosis can be confirmed by whole blood DNA analysis that will detect known mutations in roughly 80% of patients.11 The plasma acylcarnitine profile serves as a screening test that will most often detect CPT II deficiency when the patient is symptomatic; however, functional enzyme analysis of fibroblasts or muscle tissue presumably offers higher sensitivity.10
Patients with CPT II deficiency should be instructed to avoid prolonged fasting (i.e., longer than 10 hours). Sustained, intensive exercise should be avoided. Carbohydrate loading prior to and during exercise may prevent attacks. We advocate dietary supplementation with medium-chain triglycerides, which provides an alternative substrate for fatty acid oxidation involving long-chain fatty acids.12
Mitochondrial Oxidative Phosphorylation Disorders
Defects of mitochondrial oxidative phosphorylation (mitochondrial disorders; respiratory chain disorders) are typified by multisystem involvement frequently involving muscle.1 Not infrequently, though, mitochondrial disorders manifest as isolated myopathies, as opposed to the encephalopathy and multisystem involvement associated with the classical infantile presentation. Late-onset mitochondrial myopathies feature proximal muscle weakness, easy fatigueability, and variably elevated CK levels. Lactic acidemia is often absent. Thus, mitochondrial myopathies are frequently misdiagnosed as inflammatory myositis upon presentation.
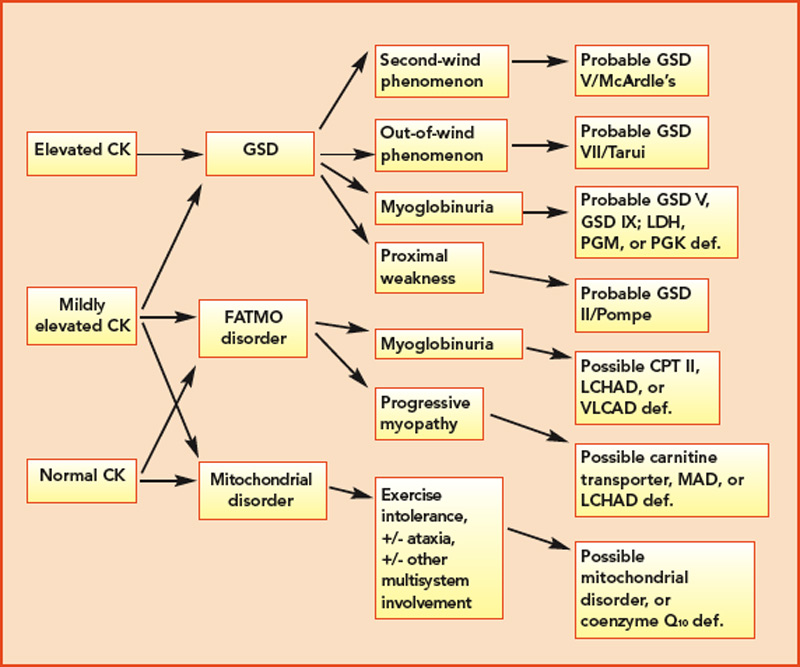
Classification by the underlying gene defect is complicated by tremendous genetic heterogeneity, with approximately 20% of mutations affecting mitochondrial DNA and the remainder affecting numerous nuclear genes. Utilizing the modified Walker criteria is essential to confirm the diagnosis of a mitochondrial myopathy because the presence of both characteristic clinical symptoms and diagnostic laboratory testing are required for confirmation of affected status.13 Muscle histology may feature ragged red fibers and decreased cytochrome C oxidase staining, or it may be normal. Functional enzymology on flash-frozen muscle tissue or fibroblasts allows quantitative assessment of electron transport chain (ETC; respiratory chain) complex activities. Flash-frozen muscle samples offer greater sensitivity than skin fibroblasts, but special handling required to maintain sample integrity is cumbersome and frequently delays or prevents the diagnosis. Noninvasive testing can be useful in some cases (see Table 3, p. 23, and Figure 1, p. 14).
Case 3
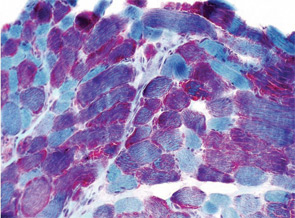
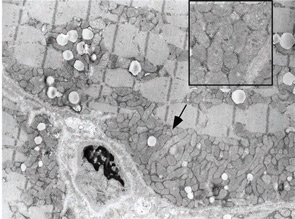
A 7-year-old male presented for inpatient consultation with recurrent vomiting, metabolic acidosis, a 6-month history of exercise intolerance accompanied by weakness, and poor weight gain. Physical examination revealed hepatomegaly and significant weakness of the lower extremities. Initial laboratory evaluation revealed significant metabolic acidosis, elevated plasma lactic acid (5 mmol/L; normal, 0.6–2.5), elevated serum CK (700 U/L; normal, 30–220), and decreased serum glucose (to 50 gm/dL). Urine organic acid analysis revealed elevated lactic acid and ketones. Plasma carnitine and acylcarnitine profiles were normal. Enzyme testing of cultured fibroblasts revealed normal carnitine palmitoyltransferase II activity and normal ETC complex II–IV activities. Subsequently, a muscle biopsy was performed, and histopathology revealed prominent ragged red fibers (see Figure 2A, p. 15). Electron microscopy revealed marked accumulation of abnormal mitochondria (see Figure 2B, p. 15). ETC testing of flash-frozen muscle confirmed a significant deficiency of complex III. Oral supplementation with coenzyme Q10 (120 mg), creatine monohydrate (3 gm), and alpha-lipoic acid (300 mg) twice daily was recommended, and gastrostomy tube feeding was initiated to provide adequate calories for growth. Within 6 months, the patient had recovered to his baseline level of activity and was gaining weight.
Mitochondrial Myopathies
Mitochondrial myopathies are emerging as a more frequent cause of metabolic myopathy than previously recognized. Classically, the mitochondrial myopathies were associated with point mutations in tRNA genes or large deletions involving the mitochondrial genome (e.g., myoclonic epilepsy and ragged red fibers or Kearns-Sayre syndrome). More recently, the deficiency of coenzyme Q10 synthesis has emerged as a cause for isolated myopathy.2 Interestingly, the underlying gene defect in cases of isolated myopathy has involved the electron transferring protein dehydrogenase (ETFDH) gene that was previously described in patients with multiple acyl-CoA dehydrogenase deficiency (MAD deficiency; glutaric acidemia type II).14 Previous reports attributed MAD deficiency to a “trifunctional protein deficiency,”1 creating confusion with a well-recognized trifunctional protein deficiency that affects three enzyme functions in FAO including LCHAD.10
Diagnosing coenzyme Q10 deficiency myopathy provides the great benefit of confirming the value of high-dose coenzyme Q10 supplementation. Establishing the diagnosis requires assaying coenzyme Q10 activity in flash-frozen muscle (see Table 3, above). In general, the mitochondrial myopathies are most effectively diagnosed via studies on flash-frozen muscle tissue, which will demonstrate abnormalities on ETC testing.
An Approach to the Diagnosis of Metabolic Myopathies
Aggressively pursuing the diagnostic evaluation of patients with a suspected metabolic myopathy is justified by the potential for therapeutic intervention and a need to provide the patient with prognostic information and appropriate genetic counseling. Proper diagnosis also avoids unnecessary testing and inappropriate therapies such as immunomodulatory therapy for a suspected inflammatory process. A carefully designed evaluation, guided by clinical and laboratory information, will allow proper diagnosis with minimally invasive testing (see Figure 1, p. 14).
For example, the presentation of a patient with recurrent rhabdomyolysis and second-wind phenomenon should prompt blood testing for mutations underlying McArdle’s disease. In contrast, exercise intolerance accompanied by weakness that is exacerbated by carbohydrate intake (out-of-wind phenomenon) suggests GSD type VII (PFK deficiency) that might be confirmed through gene sequencing or muscle-enzyme testing. At times, reaching the diagnosis will require more invasive testing, including histology, histochemistry, and enzyme analysis of muscle tissue, such as in the mitochondrial myopathies (see Table 3, above). Establishing the diagnosis of CPT II deficiency frequently requires enzyme testing of cultured fibroblasts because mutation testing often identifies only a single mutation.
Progressing from careful clinical evaluation to the least invasive testing likely to yield a diagnosis and finally to the muscle biopsy when necessary is a reasonable strategy. Several biochemical genetics laboratories offer “myoglobinuria panels” that can facilitate enzyme testing. If the decision is made to proceed to muscle biopsy, the importance of careful planning to ensure adequate sampling, processing, and preservation cannot be overstated. Collaboration with all of the consultants involved, including the rheumatologist, metabolic specialist, neurologist, surgeon, primary physician, and biochemical laboratory director under the direction of an identified team leader will enhance the chances of establishing a correct diagnosis in a timely, cost-effective manner.
Acknowledgements
We wish to thank our patients and colleagues for their support and understanding while we developed the concepts underlying this paper. We thank Dr. Edward Bossen at Duke University for images and Dr. John Shoffner at Medical Neurogenetics for comments.
Dr. Smith is a medical instructor in the division of pediatric neurology, department of pediatrics at Duke University Medical Center in Durham, N. C. Dr. Koeberl is associate professor of pediatrics and molecular genetics and microbiology at Duke University Medical Center.
References
- Wortmann RL, DiMauro S. Differentiating idiopathic inflammatory myopathies from metabolic myopathies. Rheum Dis Clin N Am. 2002;28:759-778.
- Quinzii CM, Hirano M, DiMauro S. CoQ10 deficiency diseases in adults. Mitochondrion. 2007;7 Suppl:S122-S126.
- The SEARCH Collaborative Group, Link E, Parish S. SLCO1B1 variants and statin-induced myopathy—a genome-wide study. New Eng J Med. 2008;359:789-799.
- Genetic risk factors associated with lipid-lowering drug-induced myopathies. Muscle Nerve. 2006;34:153-162.
- Chen YT. Glycogen storage diseases. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Eds. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001:1521-1551.
- DiMauro S, Lamperti C. Muscle glycogenoses. Muscle Nerve. 2001;24:984-999.
- Haller RG. Treatment of McArdle disease. Arch Neurol. 2000;57:923-924.
- Lucia A, Nogales-Gadea G, Perez M, et al. McArdle disease: What do neurologists need to know? Nat Clin Pract Neurol. 2008;4:568-577.
- Andersen ST, Haller RG, Vissing J. Effect of oral sucrose shortly before exercise on work capacity in McArdle disease. Arch Neurol. 2008;65:786-789.
- Rinaldo P, Matern D, Bennett MJ. Fatty acid oxidation disorders. Annu Rev Physiol. 2002;64:477-502.
- Vladutiu GD. The molecular diagnosis of metabolic myopathies. Neurol Clin. 2000;18:53-104.
- Gillingham MB, Scott B, Elliott D, Harding CO. Metabolic control during exercise with and without medium-chain triglycerides (MCT) in children with long-chain 3-hydroxy acyl-CoA dehydrogenase (LCHAD) or trifunctional protein (TFP) deficiency. Mol Genet Metab. 2006;89:58-63.
- Scaglia F, Towbin JA, Craigen WJ, et al. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics. 2004;114:925-931.
- Gempel K, Topaloglu H, Talim B, et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain. 2007;130:2037-2044.
- Orngreen MC, Schelhaas HJ, Jeppesen TD, et al. Is muscle glycogenolysis impaired in X-linked phosphorylase b kinase deficiency? Neurology. 2008;70:1876-1882.


