The complex nature of the various metabolic pathways involved is reflected in the broad phenotypic presentation of many of these disorders. Classic symptoms of the metabolic myopathies, including exertional fatigue, myalgias, cramping or contractures, and myoglobinuria may emerge during times of high energy demand due to exercise, illness, or fasting. The clinical presentation of these disorders is influenced by the metabolic step involved because the predominant energy substrate varies as a function of the intensity and duration of exercise. Glycogen breakdown provides substrates for cellular energy production during rapid, vigorous exercise, whereas FAO is the main player during prolonged exercise and fasting. At rest, free fatty acids provide the major skeletal muscle energy source via aerobic metabolism. Brief, high-intensity (isometric) exercise such as weightlifting relies heavily on anaerobic glycogenolysis for a rapid energy supply. With increasing duration of exercise, glycogen stores are depleted, and muscle fatigue emerges. The primary source of energy for muscle for light- to moderate-intensity exercise of short duration is predominantly glucose and free fatty acids. As exercise duration increases, so does the demand for free fatty acid–derived energy. This explains the characteristic pattern of cramping, stiffness, and myalgias shortly after brief, maximal exertion associated with several of the glycogen storage diseases (GSDs). In contrast, symptoms associated with disorders of lipid metabolism typically appear after prolonged, moderate-intensity exercise, such as jogging or swimming. Thus, patients with McArdle’s disease (glycogen storage disease type V; GSD V) become symptomatic during intense exercise, whereas carnitine palmitoyltransferase II (CPT II) deficiency, an FAO disorder, presents following sustained exercise and other metabolic stressors.
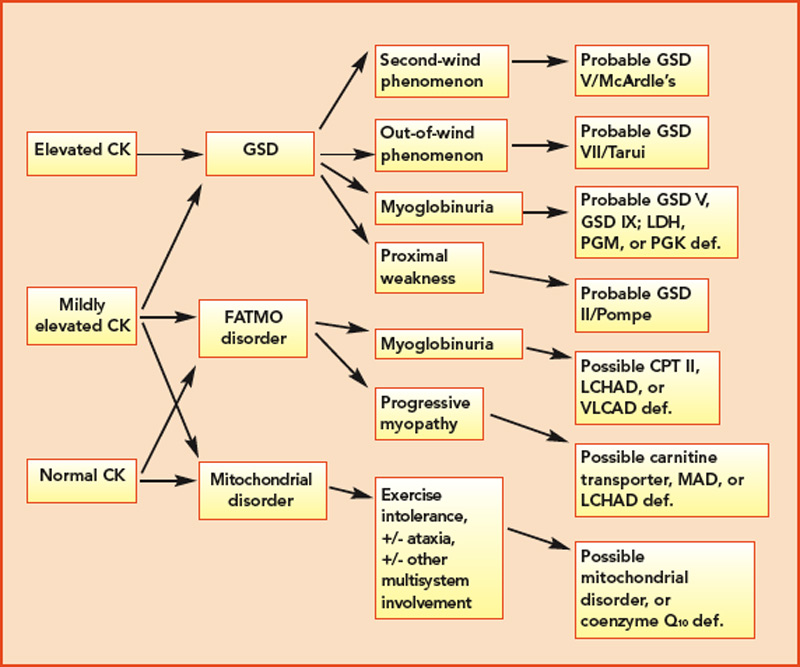
The variable and often transient nature of these symptoms can present a challenging diagnostic dilemma. Depending on the underlying defect, some patients may present in infancy or early childhood, whereas others may not present until well into adulthood. Some disorders may be strictly limited to muscle pathology, whereas others, particularly those involving mitochondrial dysfunction, may demonstrate multisystem involvement. Furthermore, a number of acquired and inherited conditions may mimic metabolic myopathies and must be properly diagnosed both to allow appropriate treatment and counseling and in order to avoid unnecessary and potentially harmful treatments (see Table 2, p. 23). Recently, drug therapy for hypercholesterolemia with statins has emerged as a cause for iatrogenic myalgia.2 The mechanism of muscle injury is not known, but there appears to be a dose-related effect, with higher statin doses more frequently leading to myopathy and rhabdomyolysis. A recent study has also associated an increased risk for statin-induced myopathy in patients harboring a common single-nucleotide polymorphism in the SLCO1B1 gene on chromosome 12, which is related to hepatic uptake of statin drugs.3 The incidence of alleles for rare metabolic myopathies, either in homozygotes or heterozygotes, was increased in a large group evaluated for statin-induced myopathy, and muscle coenzyme Q10 deficiency was highly prevalent in that group.4 Whether the coenzyme Q10 deficiency responds to supplementation remains to be established, but the biochemical abnormality could represent an important lead regarding the pathogenesis of statin-induced myopathy.


