Patients with CPT II deficiency should be instructed to avoid prolonged fasting (i.e., longer than 10 hours). Sustained, intensive exercise should be avoided. Carbohydrate loading prior to and during exercise may prevent attacks. We advocate dietary supplementation with medium-chain triglycerides, which provides an alternative substrate for fatty acid oxidation involving long-chain fatty acids.12
Mitochondrial Oxidative Phosphorylation Disorders
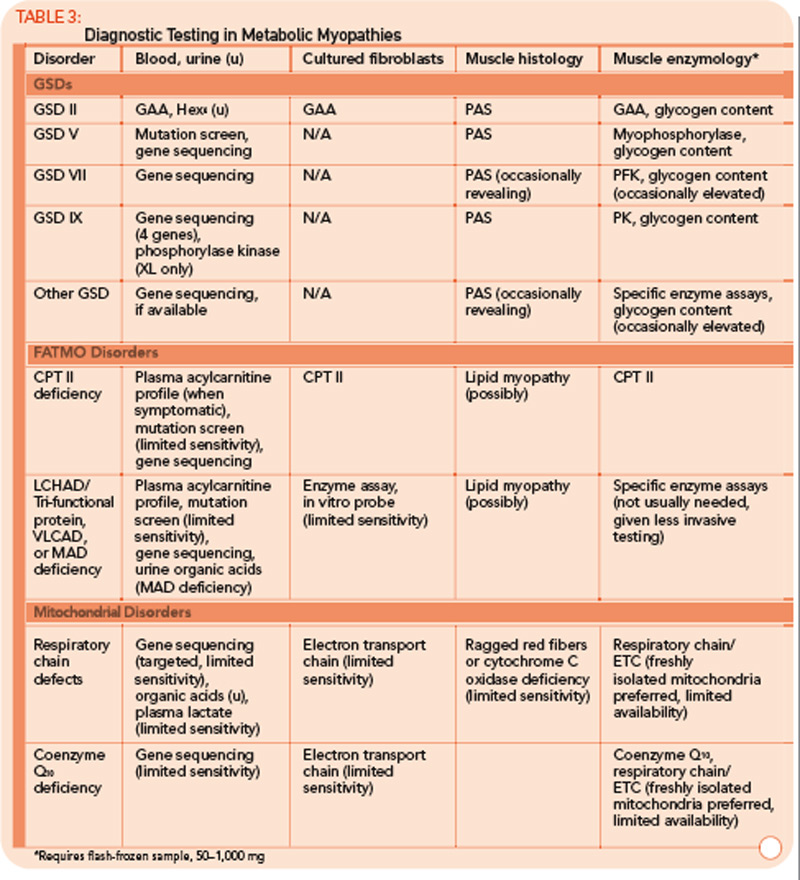
Defects of mitochondrial oxidative phosphorylation (mitochondrial disorders; respiratory chain disorders) are typified by multisystem involvement frequently involving muscle.1 Not infrequently, though, mitochondrial disorders manifest as isolated myopathies, as opposed to the encephalopathy and multisystem involvement associated with the classical infantile presentation. Late-onset mitochondrial myopathies feature proximal muscle weakness, easy fatigueability, and variably elevated CK levels. Lactic acidemia is often absent. Thus, mitochondrial myopathies are frequently misdiagnosed as inflammatory myositis upon presentation.


