
Gout is arguably the best understood rheumatic disease. The diagnosis of gout is typically straightforward, and effective serum urate–lowering drugs and anti-inflammatory compounds are available to manage the majority of patients with gout.There has been a sense of complacency about gout—until recently. The last two decades have seen a remarkable resurgence of gout in the United States. This comeback has clinicians facing increasingly complex cases in which age, comorbidities, and concomitant medications impose difficult management decisions.
Further, physicians are increasingly limited by the lack of new Food and Drug Administration (FDA)–approved therapeutic options for gouty inflammation and hyperuricemia management for patients who present with the key elements of difficult gout. (See Table 1)
Increased Prevalence and Clinical Complexity
Over the last two decades in the United States, the prevalence of gout has approximately doubled. A perfect storm of factors has drived increases in gout prevalence and clinical complexity. The graying of the population as a whole is a central factor because the greatest rise in gout prevalence by far has been among the aged. This situation is clinically problematic because polypharmacy, significant comorbidities, and altered drug disposition and toxicity profiles are common problems in the elderly. The wide use of diuretics in association with underlying chronic renal insufficiency and/or congestive heart failure has rendered difficult gout partly an iatrogenic problem in many patients. The clinical character of tophaceous gout is changing, such that the elderly female with chronic polyarticular gout presents a common problem in the clinic and for consultative practice of inpatient rheumatology. (See Figure 1)
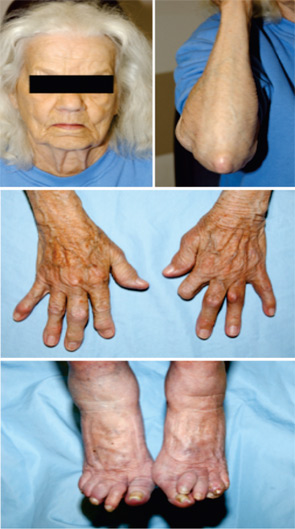
Factors confluent with aging that are changing the prevalence and character of gout include the marked increased prevalence of hypertension (with associated thiazide diuretic use); altered dietary trends; and the rising prevalence of obesity, metabolic syndrome, and end-stage renal disease over the last few decades.1 Improved survival in coronary artery disease, congestive heart failure, and diabetes mellitus, and broad use of loop diuretics (and possibly low-dose acetylsalicylic acid as well) also appear to contribute to the expanded pool of subjects with chronic, severe hyperuricemia. Finally, the limitations of allopurinol therapy render gout management difficult in a sizeable fraction of patients, particularly those with significant renal insufficiency or uric acid overproduction where uricosuric therapy is not a viable alternative to allopurinol. (See Table 2)
Limitations of Allopurinol
Allopurinol is primarily a pro-drug that must be intestinally absorbed and then converted in the liver to the active metabolite oxypurinol. The half-life of oxypurinol is largely determined by renal elimination and is normally 24 hours. Oxypurinol accumulates in association with diminished glomerular filtration rate. Notably, severe hypersensitivity to allopurinol appears partially dose-dependent. Major allopurinol hypersensitivity syndrome associated with multi-organ system damage has a mortality rate of approximately 20%. Fortunately this problem has been uncommon (i.e., no more than 100 reported cases). Nevertheless, a drug rash—typically preceded by pruritus—occurs in about 2% of allopurinol-treated patients. Only a half of these subjects (at most) successfully resumes maintenance allopurinol after oral desensitization.
Pharmacogenomics of severe cutaneous adverse reactions to allopurinol in Han Chinese are such that the odds ratio is 580:1 in HLA-B*5801–positive subjects.2 However, it is not known if this finding holds for other populations. Stepwise upward adjustment of starting allopurinol doses based on creatinine clearance is common and acceptable medical practice to dampen the risk of severe allopurinol hypersensitivity. Yet there is no precise method to predict or prevent allopurinol hypersensitivity in the population at large. Moreover, optimal dosing of allopurinol remains controversial—particularly in patients with chronic renal insufficiency.
Concern about precipitating major allopurinol hypersensitivity syndrome often drives practitioners to use allopurinol maintenance doses that are too low for many patients (specifically, doses less than or equal to 300 mg/day in patients with preserved renal function and 100–200 mg/day in patients with creatinine clearance less than 60 mL/min), which probably enlarges the pool of refractory gout. The reported patient compliance with maintenance allopurinol is around 50%, indicating a systematic failure of gout patient education by practitioners.
Precipitation of acute gout flares is a common form of allopurinol intolerance, but can be satisfactorily addressed in most patients by prophylactic colchicine with evidence-basis, or by prophylactic non-steroidal anti-inflammatory drugs (NSAIDs without evidence-basis at this time. However, allopurinol intolerance—manifesting, for example, as minor gastrointestinal and central nervous system toxicity—occurs in up to 10% of patients. Allopurinol hepatotoxicity typically presents as elevation in transaminases, a manifestation that can be difficult to precisely attribute to allopurinol. Unfortunately, more severe hepatotoxicity can develop, often cryptically. The nonselectivity of allopurinol effects in both purine and pyrimidine metabolism potentially factors into gastrointestinal, central nervous system, and hematologic toxicities.
Advances: URAT1 and Uricosurics
Hyperuricemia in most patients with both primary and secondary gout is driven by renal underexcretion of uric acid. Stunning advances have occurred in the understanding of uric acid handling in the nephron. Urate anion exchange with organic anions at the apical side of renal proximal tubule epithelial cell by URAT1 (SLC22A12)—a member of the organic anion transporter (OAT) family—is the linchpin for renal reabsorption of urate anion from the tubular lumen.
The work of Hitoshi Endou, MD, PhD, and colleagues has also revealed that the particularly potent and effective uricosuric benzbromarone, as well as several less potent uricosurics (probenecid, losartan, sulfinpyrazone, and high doses of salicylates), exert uricosuric activity via inhibition of URAT1.3 Further, testosterone increases and estrogen decreases URAT1 expression. Certain URAT1 single nucleotide polymorphisms (SNPs) have been linked to altered susceptibility to gout in different cohorts. Indeed, the ability to pharmacogenomically assess for potential responsiveness to specific uricosurics is close at hand.
The identification of URAT1 functions, including URAT1 association in macromolecular complexes that jointly influence sodium and urate reabsorption, provides a platform to rationally design a new generation of more selective, and potentially more effective, uricosurics. However, attendant risks of urolithiasis and renal toxicity limit the potential of more potent, rationally designed uricosurics to be clinically safe agents in managing the most needy subjects with difficult gout. Many patients with difficult gout are intolerant of allopurinol but cannot receive uricosurics as an alternative due to renal insufficiency or uric acid overproduction.
Novel Xanthine Oxidase Inhibitors
Xanthine oxidase is reversibly interconvertible between reduced and oxidized forms (xanthine dehydrogenase and xanthine oxidase, respectively)—both of which catalyze the formation of uric acid as the end product of purine nucleotide catabolism.4 (See Figure 2) However, oxypurinol dissociates from—and does not effectively inhibit—uric acid formation by the oxidized form of the enzyme. Though oxypurinol is tolerated in some allopurinol-hypersensitive patients, oxypurinol carries many of the limitations of allopurinol, is variably absorbed in the gut, and has not yet performed as a satisfactorily effective antihyperuricemic agent in adequately powered clinical trials. One wonders if xanthine oxidase may exist predominantly in the oxidized form in some patients where robust doses of allopurinol fail to satisfactorily lower serum uric acid. In my clinical practice, I order serum oxypurinol levels for patients in whom there are questions about either compliance with allopurinol or unidentified pharmacogenomic limitations to allopurinol treatment.
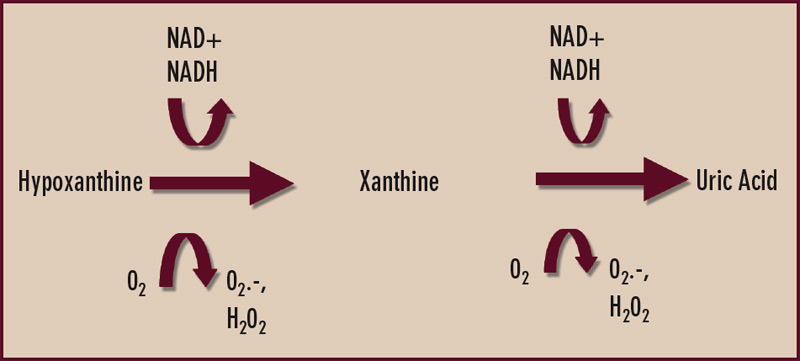
Given the aforementioned limitations of allopurinol, there is interest in selective xanthine oxidase inhibition for hyperuricemia using drugs without a purine-like backbone. One such drug, febuxostat, selectively occupies the substrate access channel to the catalytic site of xanthine oxidase. Significantly, febuxostat inhibits both the reduced and oxidized forms of xanthine oxidase and does not affect interconversion of the enzyme from one form to the other. In a large, randomized, phase III study of gout patients by Michael Becker, MD, and colleagues, febuxostat at doses of 80 and 120 mg daily was more effective than standard dosing (300 mg daily) of allopurinol in reducing serum urate to a target level of less than 6 mg/dL over one year of treatment.5 Febuxostat remains under FDA review, and detailed assessment of febuxostat in patients with renal insufficiency is expected.
A serum urate level of less than 6 mg/dL is widely advocated as an ideal therapeutic target because it is well below the theoretic limit of urate solubility in human tissues of between 6.7 and 7.0 mg/dL, the benchmark provided by in vitro studies in physiologic buffers and plasma. In response to 80 mg and 120 mg of febuxostat daily, the mean drop in serum urate from baseline to the final visit was 44.7% and 51.5%, respectively.5 The majority of allopurinol-treated patients (approximately two-thirds) did not attain sustainable serum urate lowering to less than 6 mg/dL at the final visit.
An important finding in the febuxostat phase III trial was that 300 mg allopurinol daily lowered mean serum urate by a mean of approximately 33%.5 Hence, the average patient with a serum urate of less than or equal to 9 mg/dL should achieve ideal serum-urate lowering with the typical daily dosage of allopurinol. In my experience, higher doses of allopurinol can safely achieve optimal serum-urate lowering in many additional patients. An additional finding in the febuxostat study was that the frequency of gout flares at the end of the study period was comparable in febuxostat- and allopurinol-treated patients, despite the more potent serum-urate lowering by febuxostat. Indeed, serum-urate lowering to less than the “ideal” target level of less than 6.0 mg/dL (but to levels between 6.0 and 6.7 mg/dL) may be sufficient to improve the course of gout in many patients.
Febuxostat-induced serum-urate lowering, like that due to allopurinol, was associated with a surge in flares of gouty arthritis when prophylactic colchicine was withdrawn only eight weeks into the therapy after the washout period.5 I advocate starting low-dose prophylactic colchicine two weeks before initiation of urate-lowering therapy. I also advocate maintaining the colchicine regimen for six months in the average patient, and longer in patients with persistent tophi. More intensive serum urate–lowering therapy has the potential to induce more active tophus remodeling, a state that is likely pro-inflammatory at tophaceous deposits.
Uses for Uricase
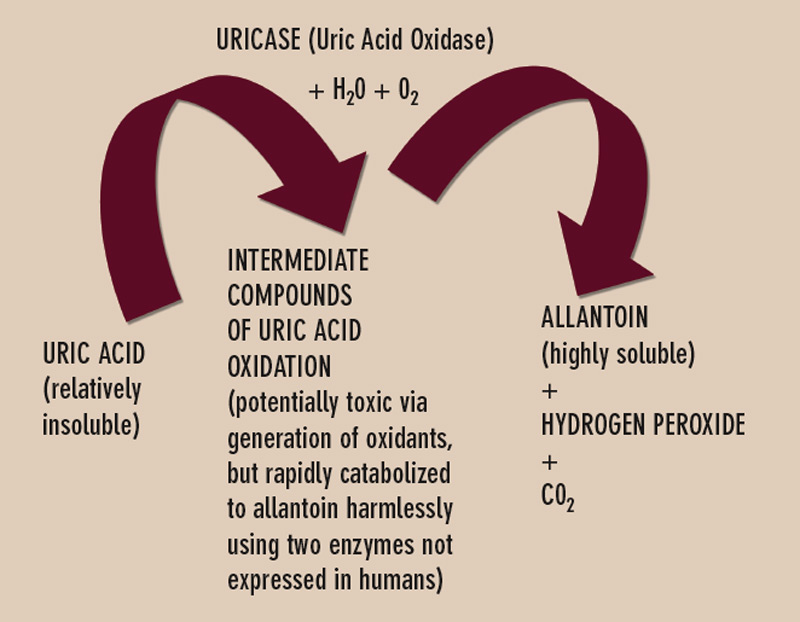
Uricase is one of several enzymes of the final purine degradation pathway in nonprimate mammals and lower primates that catalyzes the conversion of relatively insoluble uric acid to highly soluble allantoin. (See Figure 3) Uricase gene silencing in evolution has rendered the human urate balance delicate. Normal rodents, for example, have a serum urate level of about 1 mg/dL, which is approximately sixfold less than that of normal humans. Humans’ lack of uricase expression may protect them from oxidative stress such as neuronal toxicity of nitric oxide–derived oxidants. Uric acid is the major circulating antioxidant. It was recently demonstrated that the oxidation of uric acid by uricase produces reactive and potentially toxic intermediates in lower mammals that are rapidly degraded by two enzymes that humans also fail to express. Further, the slow, spontaneous conversion of uric acid to allantoin yields the oxidant hydrogen peroxide as a byproduct.
Currently, the recombinant fungal uricase rasburicase is FDA approved for abbreviated, single-course use to prevent tumor lysis syndrome in subjects with hematologic malignancy. This recombinant uricase is neither humanized nor modified by PEGylation. Thereby it is particularly immunogenic and limited by a relatively short half-life. Side effects, including anaphylactic reactions, have been limiting. Robust amounts of neutralizing antibodies to uricase can also develop. PEGylation of uricase promotes less immunogenicity and a longer half-life, and appears better suited for urate-lowering therapy in refractory gout. Indeed, PEG-ylated uricase preparations have induced profound serum urate–lowering in clinical trials. The shift from subcutaneous to intravenous administration of PEGylated porcine recombinant uricase (Puricase) reduced hypersensitivity reactions.6 However, immunogenicity could still constrain safety and longer-term efficacy of current PEGylated uricase preparations in clinical development.
Oxidative stress via uricase-induced hydrogen peroxide generation is another potential concern with enzyme therapy use. Erythrocyte-borne catalase should theoretically be sufficient to degrade circulating hydrogen peroxide generated via typical doses of uricase. Nevertheless, latent vulnerability to oxidative stress with uricase administration is indicated by hemolysis and methemoglobinemia as classic uricase side effects in patients with G6PD deficiency. In my opinion, the primary therapeutic niche for uricase in gout will be limited duration treatment (months) as induction therapy to debulk macroscopic tophi in carefully selected patients with extensive disease refractory to other treatments.
Inflammation in Difficult Gout
Difficult gout includes patients who are intolerant of, or refractory to, standard therapeutic approaches to gouty inflammation: NSAIDs (or alternatively COX-2 selective inhibitors), systemic or intra-articular glucocorticosteroids, adrenocorticotropic hormone (ACTH), or colchicine.7 Clinicians can carefully adjust colchicine doses in line with renal function and employ combinations of low-dose colchicine with other anti-inflammatories to manage some of these subjects with renal or hepatic dysfunction, gastrointestinal comorbidity, poorly controlled diabetes mellitus, age, and post-operative state.7 However, polyarticular gout flares refractory to therapy do occur. A striking example is seen in major organ–transplant recipients receiving cyclosporine who develop severe gouty arthritis despite maintenance low-dose prednisone. Some patients have major gout flares when starting or adjusting uric acid–lowering therapies, including the intensive serum urate–lowering regimens currently under clinical development.
The primary pathologic hallmark of acute gout is neutrophil influx into the joint fluid, consistent with interleukin 1 (IL-1)–induced and tumor necrosis factor-a (TNF-a)–induced activation of endothelium and with tissue generation of neutrophil chemoattractants that drive and sustain the inflammation. Certain molecular mechanisms of action of anti-inflammatory agents used in gout could be exploited for difficult gout. For example, the rapid onset of the therapeutic action of ACTH in gout appears mediated by binding to the melanocortin receptor 3 (MR3) on phagocytes in the periphery. Stimulation of MR3 by itself can suppress experimental gouty inflammation.8 We also know that low-dose prophylactic colchicine prevents gouty inflammation in part by inhibiting redistribution the neutrophil adhesion molecule E-selectin in vascular endothelium.
Primitive Innate Immune Response
Acute gout is a prototype of the primitive innate immune “early induced” response, with the characteristic recurrent, paroxysmal nature of gouty arthritis representing failure of monosodium urate crystals to directly induce durable protective immunity.9 Uptake of both apoptotic neutrophils and urate crystals by resident and recruited “professional phagocytes” mobilizes a rapid effort to quell the inflammation.9,10 The recent elucidation of new molecular biologic mechanisms of gouty inflammation may light the way to novel therapeutics.
Innate immunity against invading pathogens involves the distribution of pattern recognition receptors and recognition molecules outside cells, as well as on the cell surface and in the cytoplasm. Innate immune responses to urate crystals that drive gouty inflammation include extracellular crystal-induced complement membrane attack complex assembly, which promotes the CXC chemokine receptor 2 (CXCR2)–binding chemokine expression essential for the neutrophil influx that drives gouty inflammation.9,11,12 (See Figure 4)
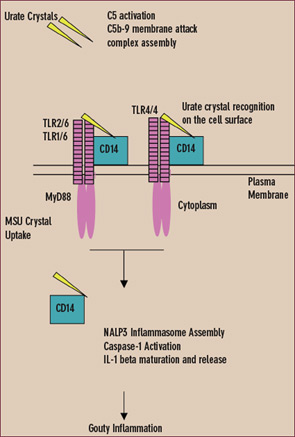
Experimental gouty inflammation is triggered by IL-1 receptor signaling by resident cells at the site of urate crystal injection.13 Macrophage recognition and uptake of urate crystals by the cell surface Toll-like receptors (TLRs) 2 and 4 and by signaling of the shared intracellular TLR adaptor protein MyD88 drives both pro- and anti-inflamamtory cytokine expression.14 Depending on the locus of experimental urate crystal injection, TLR2 and TLR4 expression determines dominant anti-inflammatory effects (such as induction of transforming growth factor b) or pro-inflammatory effects in vivo. However, MyD88, which also is a critical adaptor protein for the IL-1 receptor, is central to both urate crystal uptake by cultured macrophages and urate crystal-induced inflammation in vivo.13,14
In the cytoplasm, urate crystals also employ the cytoplasmic pattern recognition receptor NALP3 (cryopyrin) to drive inflammation.11,12 Upon ligand recognition, NALP3, like other members of the nucleotide-binding oligomerization domain (NOD)–like receptor family, assembles with other proteins in large complexes termed inflammasomes. When NALP3 complexes with caspase-recruiting domain (CARD)–carrying adaptor proteins, activation of the latent inflammatory mediator caspase-1 is stimulated. The activated NALP3 inflammasome complex thereby induces proteolysis of pro–IL-1b and subsequent maturation and release of IL-1b.11
Like TLRs, NALP3 contains leucine-rich repeats.15 Leucine-rich repeat domains of TLRs recognize various pathogen-associated molecular patterns. Therefore, the leucine-rich repeat domain of NALP3 is believed to directly sense components of microbial and other pathogens, stress signals (e.g., high amounts of adenosine triphosphate [ATP] released from dying cells), and urate crystals. Extracellular urate crystal binding of the CD14, an adaptor protein shared by TLR2 and TLR4, does not alter phagocytosis of urate crystals by macrophages, but CD14 switches macrophage encounters with urate crystals that are non-inflammatory into pro-inflammatory events.16 CD14 does so by allowing urate crystals to fully engage the NALP3 inflammasome to activate caspase-1 and induce release of mature IL-1b.16
The last two decades have seen a remarkable resurgence of gout in the United States. This comeback has clinicians facing increasingly complex cases in which age, comorbidities, and concomitant medications impose difficult management decisions.
Autoinflammatory Cryoprinopathies Identify Potential Therapeutic Targets
Over the last few years, mutations in NALP3 have been linked to certain autosomal dominant inherited auto-inflammatory syndromes of differing severity. These syndromes include familial cold-induced auto-inflammatory syndrome, Muckle-Wells syndrome, and neonatal onset multisystem inflammatory disease/chronic infantile neurological cutaneous and articular syndrome.15 The self-resolution of periodic flares of systemic inflammation in these autoinflammatory cryopyropathies is remarkably similar to the typical course of gouty arthritis, which reflects the shared central role of NALP3.11 Recent research on autoinflammatory syndromes has brought to light new mechanisms of action of the venerable gout anti-inflammatory therapeutic colchicine. Specifically, urate crystal–induced NALP3 inflammasome protein complex assembly is suppressed by high concentrations of colchicine, likely by interfering with internalization of urate crystals and thereby inhibiting crystal delivery to the inflammasome.11
Therapeutic studies of autoinflammatory cryopyrinopathies have already provided information of substantial potential value for developing novel and more selective molecular therapeutics for difficult gout inflammation. The responsiveness of familial cold-induced autoinflammatory syndrome, Muckle-Wells syndrome, and neonatal onset multisystem inflammatory disease/chronic infantile neurological cutaneous and articular syndrome to IL-1 receptor antagonism is particularly noteworthy because a preliminary, uncontrolled study of 10 gout patients suggested therapeutic benefit of anakinra. Significantly, experimental gouty inflammation in rodents appears more IL-1b driven than TNF-a driven.12 Further investigation for gouty arthritis is warranted for not only IL-1 inhibitors but also the caspase-1 inhibitors currently under clinical development for osteoarthritis treatment. The ATP-binding function of NALP3 is critical for inflammatory signaling, as is NALP3 leucine-rich repeat domain interaction with the heat shock protein HSP90.17,19 Hence, inflammation in difficult gout could be prime applications of novel therapeutics that selectively target the NALP3 ATP-binding domain or HSP90.17,18
Conclusion
Gout is increasingly common and has become more clinically complex in recent years, particularly in older patients. Clinical development continues for novel, effective hypourecimic agents to counter the limitations of the current generation of universally available primary uric acid–lowering agents for gout (allopurinol and probenecid) in difficult gout. The linkage of gouty inflammation to innate immunity transduced by the NALP3 inflammasome, and the definition of gouty inflammation to be caspase-1 and IL-1b driven (as in familial autoinflammatory cryopyrinopathy syndromes) augur well for novel treatment strategies for gouty inflammation in refractory patients.
Dr. Terkeltaub is San Diego VA rheumatology section chief and professor of medicine and rheumatology training program director at the University of California, San Diego. He thanks his lab and clinical colleagues and patients and staff at these institutions.
He is supported by research awards from the VA Research Service and National Institutes of Health and also receives research grant support from TAP Pharmaceuticals. In the last year, Dr. Terkeltaub served as a paid consultant to TAP, AR Scientific, Abbott, Regeneron, and Novartis.
References:
- Bieber JD, Terkeltaub RA. Gout: on the brink of novel therapeutic options for an ancient disease. Arthritis Rheum. 2004;50:2400-2414.
- Hung SI, Chung WH, Liou LB, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci USA. 2005;102:4134-4139.
- Anzai N, Kanai Y, Endou H. New insights into renal transport of urate. Curr Opin Rheumatol. 2007;19:151-157.
- Terkeltaub R, Bushinsky DA, Becker MA. Recent developments in our understanding of the renal basis of hyperuricemia and the development of novel antihyperuricemic therapeutics. Arthritis Res Ther. 2006;8 Suppl 1:S4. Epub 2006 Apr 12.
- Becker MA, Schumacher HR Jr, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med. 2005;353:2450-2461.
- Sundy JS, Ganson NJ, Kelly SJ, et al. Pharmacokinetics and pharmacodynamics of intravenous PEGylated recombinant mammalian urate oxidase in patients with refractory gout. Arthritis Rheum. 2007;56:1021-1028.
- Terkeltaub RA. Clinical practice. Gout. N Engl J Med. 2003;349:1647-1655.
- Getting SJ, Lam CW, Chen AS, Grieco P, Perretti M. Melanocortin 3 receptors control crystal-induced inflammation. FASEB J. 2006;20:2234-2241.
- Liu-Bryan R, Terkeltaub R. Evil humors take their toll as innate immunity makes gouty joints TREM-ble. Arthritis Rheum. 2006;54:383-386.
- Rose DM, Sydlaske AD, Agha-Babakhani A, Johnson K, Terkeltaub R. Transglutaminase 2 limits murine peritoneal acute gout-like inflammation by regulating macrophage clearance of apoptotic neutrophils. Arthritis Rheum. 2006;54:3363-3371.
- Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237-241.
- So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther. 2007;9:R28 [Epub ahead of print].
- Chen CJ, Shi Y, Hearn A, et al. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest. 2006;116:2262-2271.
- Liu-Bryan R, Scott P, Sydlaske A, Rose DM, Terkeltaub R. Innate immunity conferred by Toll-like receptors 2 and 4 and myeloid differentiation factor 88 expression is pivotal to monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum. 2005;52:2936-2946.
- Aksentijevich I, Putnam CD, Remmers EF, et al. The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum. 2007;56:1273-1285.
- Scott P, Ma H, Viriyakosol S, Terkeltaub R, Liu-Bryan R. Engagement of CD14 mediates the inflammatory potential of monosodium urate crystals. J Immunol. 2006;177:6370-6378.
- Duncan JA, Bergstralh DT, Wang Y, et al. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc Natl Acad Sci USA. 2007;104:8041-8046.
- Mayor A, Martinon F, De Smedt T, Petrilli V, Tschopp J. A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat Immunol. 2007;8:497-503.


