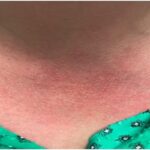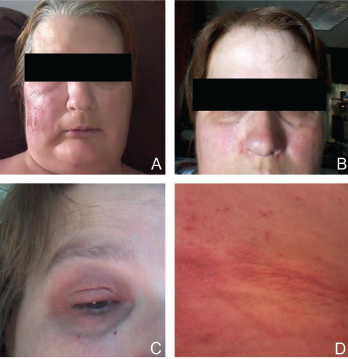
Figure 1. Patient’s rash. A and B: swollen and erythematous patches with maculopapular changes on the face; C: eyelid swelling with discoloration; D: erythematous patches and plaques on abdominal wall.
A 51-year-old Caucasian female was referred by a local rheumatologist to the Center of Autoinflammatory Diseases at Stony Brook University, N.Y., for an unusual disease presentation. The patient had had recurrent polyarthritis, fever and rash for the previous three years. She described having a migratory polyarthritis affecting the shoulders, knees, ankles and bilateral forefoot, with swelling and variable morning stiffness.
The patient also reported recurrent fever that reached a maximum of 102°F and rash. Febrile episodes were self-limiting, lasting for 24–48 hours and recurred three or four times a year, often around the same dates in April and August. The fever was accompanied by erythematous and swollen patches on her face, eyelid swelling with discoloration and hive-like patchy erythema on her abdomen and gluteal regions (see Figure 1). The episodes of rash often lasted 24–72 hours, with spontaneous resolution.
The physical examination showed obesity with body mass index of 35.0 kg/m² without active rash, synovitis or abdominal tenderness. Otherwise, it was unremarkable.
A thorough workup during the disease course was normal or negative for complete blood counts, comprehensive metabolic panel and urinalysis. Her erythrocyte sedimentation rate was 45 mm/hour (normal<30 mm/hour). Serologic markers for systemic autoimmune diseases were negative, including anti-nuclear antibodies, anti-ENA (Sm, RNP, SSA and SSB) and anti-dsDNA antibodies, Complement 3 and 4, rheumatoid factor, anti-cyclic citrullinated peptide antibodies, anti-neutrophil cytoplasmic antibodies, and HLA-B27. Serum 14-3-3 protein was negative and VECTRA DA score was 51 (high 45–100). Infectious workup was negative for viral hepatitis panel and Lyme test.
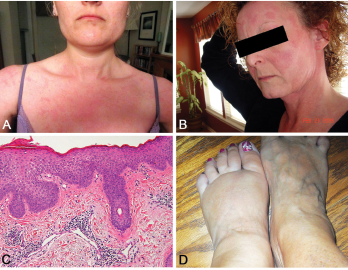
Figure 2. A and B: erythematous patches and plaques; C: spongiotic dermatitis (HE staining, 20X: epidermal hyperplasia with spongiosis and overlying hyperkeratosis and parakeratosis, as well as perivascular lymphocytic infiltrate); D: distal extremity swelling.
Other tests were normal, including thyroid-stimulating hormone, parathyroid hormone and angiotensin-converting enzyme. Radiographs of hands and feet were unremarkable as was prior chest-computed tomography. She was treated for possible seronegative arthritis with only nonsteroidal anti-inflammatory drugs without improvement.
Her clinical phenotype was suggestive of an autoinflammatory disease, so she was referred to our Center of Autoinflammatory Diseases for further workup. Due to the gastrointestinal symptoms, repeat colonoscopy showed only sigmoid diverticulosis.
Genetic testing by Next-Generation Sequencing for periodic fever panel of five genes (familial Mediterranean fever, cryopyrin-associated periodic fever syndrome, TNF-receptor-associated periodic syndrome, hyper-IgD syndrome and NOD2) detected two heterozygous variants in the NOD2 gene (IVS8+158 and 1007FS). The patient was initiated on sulfasalazine.
Discussion
Yao syndrome (YAOS, OMIM 617321), formerly called NOD2-associated autoinflammatory disease, is an autoinflammatory disease characterized by periodic fever, dermatitis, arthritis and swelling of the distal extremities, as well as gastrointestinal and sicca-like symptoms (see Figure 2, p. 44). The disorder is associated with specific NOD2 variants. YAOS is a multi-organ polygenic systemic disease and can also cause recurrent oral ulcers, pleuritis, pericarditis, lymphadenopathy, sore throat and ocular myositis. It occurs in the Caucasian population with female-to-male ratio of 2:1. Acute-phase reactants are often elevated.
YAOS differs from inflammatory bowel disease, Blau syndrome, primary Sjögren’s syndrome and monogenic hereditary systemic autoinflammatory diseases.1-3
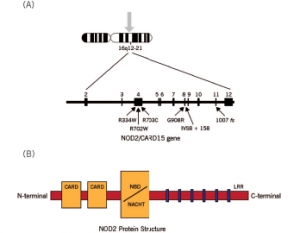
Figure 3. Schematic diagram of the NOD2 gene graph and protein structure.
The diagnosis of YAOS is dependent on the characteristic phenotype and genotype, as well as exclusion criteria (see Table 1).4 Our patient presented with typical clinical phenotype and genotype, consistent with YAOS by the diagnostic criteria.
Susceptibility to YAOS is conferred by variation in the NOD2 gene on chromosome 16q12-21 (see Figure 3).3 Genetic testing for NOD2 variants is an integral part of the disease diagnosis. The most frequent variant seen in YAOS is NOD2 IVS8+158, and up to 25% of patients can have concurrent R702W. Other NOD2 rare variants can be seen also.
Therapeutic regimen for YAOS consists of glucocorticoids and/or sulfasalazine in general. For refractory symptoms, both IL-1 and IL-6 antagonists, such as canakinumab and tocilizumab, can be tried with effectiveness in some cases. As a systemic inflammatory disease, YAOS can involve multiple organs, but with rare influence on the internal solid organs. It can cause chronic pain syndrome, fibromyalgia and even disability in some cases. Relatively common comorbidities are fibromyalgia, asthma and urinary stones as in our case.4 YAOS is relatively common when compared with monogenic systemic autoinflammatory diseases in the American adult patient population.5
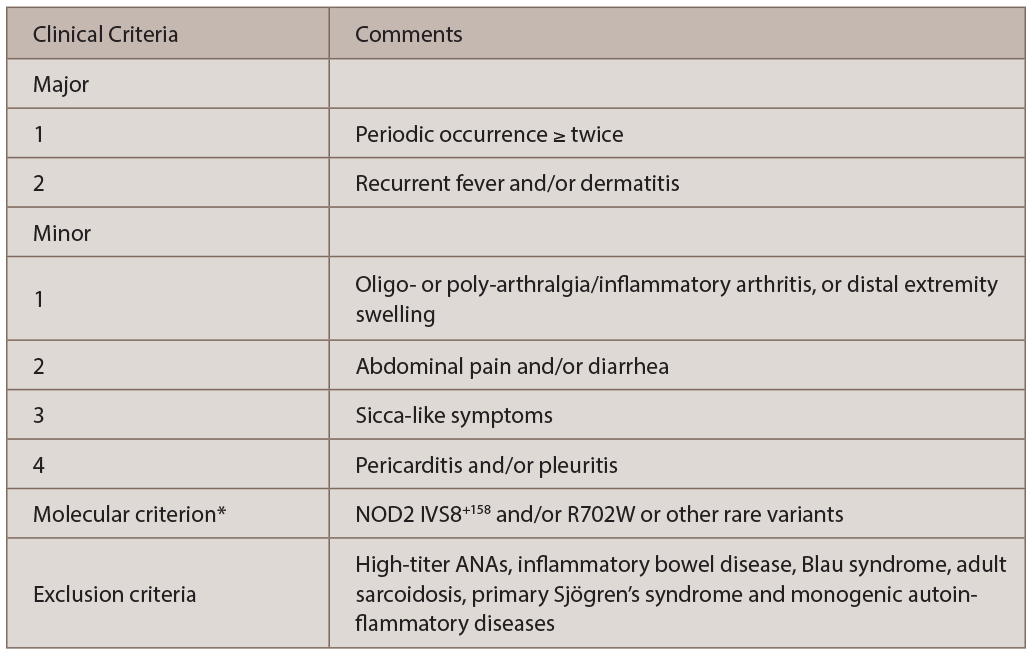
(click for larger image) TABLE 1: The Diagnostic Criteria for Yao Syndrome
The prevalence of YAOS has been estimated to be 1 to 10/100,000.3 Other medical institutions have seen or reported cases of the disease.6 Early and prompt recognition of the disease can minimize duplication of extensive and expensive testing, and with a correct diagnosis, physicians are able to manage the patients with assurance.
 Mahjabeen Haq, DO, is a rheumatology fellow in the Division of Rheumatology, Allergy and Immunology, Stony Brook University, Stony Brook, N.Y.
Mahjabeen Haq, DO, is a rheumatology fellow in the Division of Rheumatology, Allergy and Immunology, Stony Brook University, Stony Brook, N.Y.
 L. Manuela Marinescu, MD, is a rheumatologist with Rheumatology Associates of Long Island, N.Y.
L. Manuela Marinescu, MD, is a rheumatologist with Rheumatology Associates of Long Island, N.Y.
 Qingping Yao, MD, PhD, is a professor in the Department of Medicine and chief of the Division of Rheumatology, Allergy and Immunology, Stony Brook University, Stony Brook, N.Y.
Qingping Yao, MD, PhD, is a professor in the Department of Medicine and chief of the Division of Rheumatology, Allergy and Immunology, Stony Brook University, Stony Brook, N.Y.
Acknowledgment
The authors thank Mr. Chenjun Feng, Department of Medicine, Stony Brook University, N.Y., for making the composite Figure 1.
References
- Yao Q, Zhou L, Cusumano P, et al. A new category of autoinflammatory disease associated with NOD2 gene mutations. Arthritis Res Ther. 2011;13(5):R148.
- Yao Q, Su LC, Tomecki KJ, et al. Dermatitis as a characteristic phenotype of a new autoinflammatory disease associated with NOD2 mutations. J Am Acad Dermatol. 2013;68(4):624–631.
- Yao Q, Shen M, McDonald C, et al. NOD2-associated autoinflammatory disease: A large cohort study. Rheumatology (Oxford). 2015;54(10):1904–1912.
- Yao Q, Shen B. A systematic analysis of treatment and outcomes of NOD2-associated autoinflammatory disease. Am J Med. 2017;130(3):365 e13–e18.
- Yao Q, Lacbawan F, Li J. Adult autoinflammatory disease frequency and our diagnostic experience in an adult autoinflammatory clinic. Semin Arthritis Rheum. 2016;45(5):633–637.
- Estephan M, Yao Q, Springer J. Case of NOD2-associated autoinflammatory disease successfully treated with sulfasalazine. J Clin Rheumatol. 2017;23(1):58–59.