Pembrolizumab antibody, a humanized IgG4 antibody.
KATERYNA KON / Science Source
In 1888, Dr. Jan Mikulicz-Radecki reported a case of chronic, bilateral, painless enlargement of the salivary and lacrimal glands that appeared to be idiopathic.1 In subsequent years, other patients with these findings were reported, and the term Mikulicz syndrome was used to describe these cases. Although Mikulicz syndrome is now known to be associated with certain specific conditions, such as sarcoidosis and Sjögren’s syndrome, IgG4-related disease has also been identified as a potential cause.
At the 2021 ACR State-of-the-Art Clinical Symposium, John Stone, MD, MPH, professor of medicine and Edward A. Fox Chair in Medicine, Massachusetts General Hospital, Boston, provided an excellent update regarding the clinical presentation, diagnosis and management of IgG4-related disease.
The Condition
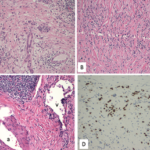
IgG4-related disease is a progressive, immune-mediated fibrotic disease that typically results in tumor-like masses in many affected organs, such as salivary and lacrimal glands, the pancreas and the kidneys. Studies have shown that CD4+ cytotoxic T lymphocytes are the predominant immune cell infiltrate in IgG4-related disease, with M2 macrophages, activated B cells and fibroblasts likely also playing a role in the generation of inflammatory masses. Classic histologic findings seen on biopsies of affected organs include lymphoplasmacytic infiltrate, obliterative phlebitis and storiform fibrosis, with a large proportion of the plasma cells staining positive for IgG4.2
Serum IgG4 concentration is a good biomarker for many patients with this condition, Dr. Stone noted. However, an elevated serum IgG4 level on its own does not clinch the diagnosis. In cases of IgG4-related disease, a high baseline serum IgG4 level can be helpful for following the disease longitudinally, because the level will often decrease with effective treatment or waning disease activity. If a patient is being treated for their IgG4-related disease and the serum IgG4 level begins to rise, clinicians should consider changing the treatment regimen.
Other biomarkers of disease may include dissociation between erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP)—with high ESR and low CRP being a typical finding in active disease—elevated IgG1 and serum IgE levels, low C3 and C4 levels, and peripheral eosinophilia.
A major advancement in the study of this condition arrived in the form of the 2019 ACR/European League Against Rheumatism (EULAR) classification criteria for IgG4-related disease.3 These criteria employ a three-step process to accurately identify patients with this disease and seek to exclude mimicking conditions.
Step 1: Identify typical involvement in at least one of the 11 possible organs typically affected by IgG4-related disease (i.e., pancreas, salivary glands, bile ducts, orbits, lacrimal glands, kidneys, lungs, aorta, retroperitoneum, pachymeninges or thyroid gland [Riedel’s thyroiditis]). Such involvement can be based on clinical or radiographic assessment, or pathologic evidence of an inflammatory process accompanied by a lymphoplasmacytic infiltrate of uncertain etiology in one of these organs.
Step 2: Apply exclusion criteria, which includes a total of 32 clinical, serologic, radiographic and pathologic items. This large array of exclusion criteria is meant to ensure patients with a wide range of autoimmune, hematologic, infectious and malignant conditions are not misdiagnosed as having IgG4-related disease. Example: Leukopenia and thrombocytopenia are unusual in IgG4-related disease, but often found in myelodysplastic syndromes, hematopoietic cancers and connective tissue diseases, such as systemic lupus erythematosus. Similarly, certain radiographic findings, such as long-bone abnormalities seen in Erdheim-Chester disease, should not be attributed to IgG4-related disease.
Step 3: Use eight weighted inclusion criteria domains, which include immunostaining of IgG positive cells, head and neck gland involvement, pancreatic and biliary tree involvement, renal involvement, chest involvement and findings of retroperitoneal fibrosis or periaortitis. In studying these classification criteria in several validation cohorts, a threshold of 20 points on the criteria scale has a sensitivity of 97–99% and a specificity of 82–85%.3
Dr. Stone explained that both a proliferative and fibrotic subtype of IgG4-related disease appear to exist. In the proliferative subtype, patients commonly experience multi-organ involvement, and typical lab findings include elevated IgG4, IgG1 and IgE levels, eosinophilia and hypocomplementemia.
Responsiveness to treatment is typically quite good in these patients. In individuals with the fibrotic subtype, patients often experience involvement of the retroperitoneum, aorta and periaortic tissue, the IgG4, IgG1, and IgE levels are typically normal or only slightly elevated, and eosinophilia and hypocomplementemia are unusual.
Additionally in the fibrotic subtype, the lymphoplasmacytic infiltrate is usually sparse, but the IgG4:IgG ratio remains greater than 40%. Response to treatment is less robust in the fibrotic than the proliferative subtype, but treatment response can still be achieved if treatment is initiated early in the disease course.4
Treatment
In Asia, treatment for IgG4-related disease typically involves 40 mg of prednisone daily for one month, followed by a taper to 5–10 mg daily and maintenance of this dose long term, Dr. Stone noted. With this treatment regimen, a high percentage of relapses occur and adverse effects from treatment remain somewhat high.
In North America and Europe, treatment usually involves 40 mg of prednisone daily for one month, followed by tapering of prednisone over 2–3 months. Although similar to the treatment approach in Asia, this regimen, unfortunately, results in frequent disease flares.
Rituximab has been used to treat multiple manifestations of IgG4-related disease, and new treatments are being explored for this patient population. 5 These agents include inebilizumab, an anti-CD19 agent; rilzabrutinib, a Bruton’s tyrosine kinase inhibitor; and elotuzumab, which targets the SLAMF7 pathway.
Many detrimental effects of IgG4-related disease come in the form of accumulated damage, causing endocrine and exocrine pancreatic failure, renal failure, hydronephrosis and the need for chronic ureteral stents, chronic hepatobiliary failure, bone-destructive sinus lesions, orbital lesions or pituitary failure. Thus, the primary goal of treatment is to halt the disease process early in its course to avoid long-term damage.
Although IgG4-related disease has only been defined in the past 20 years, Dr. Stone discussed many advances that have occurred in our understanding and management of this condition. In coming years, rheumatologists hope patients with this disease will be readily identified and treated— and perhaps, a great deal of morbidity and mortality can be prevented.
Jason Liebowitz, MD, completed his fellowship in rheumatology at Johns Hopkins University, Baltimore, where he also earned his medical degree. He is currently in practice with Skylands Medical Group, N.J.
References
- Mikulicz J. Über eine eigenartige symmetrishe Erkrankung der Thränen-und Mundspeicheldrüsen. In: Mikulicz J, editor. Beiträge zur Chirurgie: Festschrift gewidmet Theodor Billroth. Stuttgart: F.Enke; 1892. pp. 610–630. Available at: http://webapp.uibk.ac.at/alo_cat/card.jsp?id=8542696&pos=54&phys=#.
- Perugino CA, Stone JH. IgG4-related disease: An update on pathophysiology and implications for clinical care. Nat Rev Rheumatol. 2020 Dec;16(12):702–714.
- Wallace ZS, Naden RP, Chari S, et al. The 2019 American College of Rheumatology/European League Against Rheumatism classification criteria for IgG4-related disease. Arthritis Rheumatol. 2020 Jan;72(1):7–19.
- Zhang W, Stone JH. Management of IgG4-related disease. Lancet Rheumatol. 2019 Sept;1(1):e55–e65.
- Carruthers MN, Topazian MD, Khosroshahi A, et al. Rituximab for IgG4-related disease: A prospective, open-label trial. Ann Rheum Dis. 2015 Jun;74(6):1171–1177.