
esigner491 / shutterstock.com
Historically, the early approach for treating interstitial lung disease (ILD) due to systemic sclerosis (SSc) involved immunosuppressant therapy, primarily with cytotoxic agents.1 Glucocorticoids in combination with another immunosuppressant agent, such as oral azathioprine or cyclophosphamide, were often used to treat patients with severe, progressive SSc-ILD.2 However, direct evidence to support this therapeutic approach was lacking until the publication of Scleroderma Lung Study (SLS) I, a randomized, controlled trial comparing 12 months of oral cyclophosphamide with 12 months of placebo in 158 patients with relatively early SSc (<7 years from the onset of the first non-Raynaud’s symptom of SSc) and evidence of active alveolitis.3
In SLS I, patients randomized to cyclophosphamide experienced a modest improvement at a group level in the primary endpoint of the forced vital capacity (FVC) percent-predicted compared with placebo (2.53%; P<0.03) (see Figure 1).3 Moreover, patients randomized to cyclophosphamide also experienced clinically meaningful improvements in quality of life, dyspnea measures and several secondary endpoints, including total lung capacity percentage predicted and the extent of both visually assessed and computer-assisted quantitative lung fibrosis.3,4
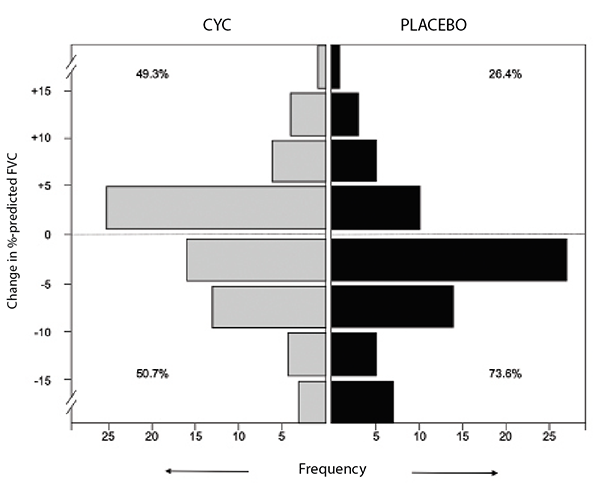
(click for larger image) Figure 1: Frequency distribution of changes from baseline to 12 months in FVC percent-predicted by treatment arm in SLS I.
However, the benefits of cyclophosphamide were tempered by safety concerns because more adverse events were observed in this study arm, including higher rates of leukopenia and neutropenia compared with placebo.3 Further, one year after treatment cessation, the FVC percent-predicted returned to baseline in both study arms. A responder analysis revealed a loss of any significant treatment effect of cyclophosphamide at 24 months for both the FVC percent-predicted and total lung capacity percent-predicted.5
Study #2
The findings of SLS I highlighted the need for additional studies to identify novel therapeutic agents for SSc-ILD with better safety profiles and more sustained treatment effects. SLS II was conceived to evaluate the safety and efficacy of daily mycophenolate mofetil, an agent that possesses both anti-fibrotic and immunomodulatory effects.6 This study compared 24 months of mycophenolate mofetil with 12 months of oral cyclophosphamide followed by 12 months of placebo in 142 patients with SSc-ILD. The entry criteria and investigating centers were nearly identical in SLS I and II.
The primary hypothesis of SLS II was that patients assigned to mycophenolate mofetil would experience significantly greater improvement in FVC percent-predicted compared with patients assigned to cyclophosphamide. This hypothesis was largely based on the assumption that continued treatment with mycophenolate mofetil for 24 months would lead to a sustained improvement in FVC percent-predicted, while cessation of cyclophosphamide at 12 months would lead to a decline in FVC percent-predicted back to baseline values at 24 months.
The final results of SLS II indicated no difference in the course of FVC percent-predicted over 24 months in patients assigned to mycophenolate mofetil vs. cyclophosphamide.6 Both treatment arms experienced significant and clinically meaningful improvements in their FVC percent-predicted over the course of the trial (see Figure 2). Sixty-five percent of those assigned to cyclophosphamide and 72% of those assigned to mycophenolate had stable or improving pulmonary function as measured by FVC percent-predicted. In addition, both treatments resulted in significant improvements in dyspnea (as measured by the transitional dyspnea index), cutaneous sclerosis and extent of radiographic fibrosis and ILD; however, no significant differences between treatment groups were observed in any of the outcome measures.
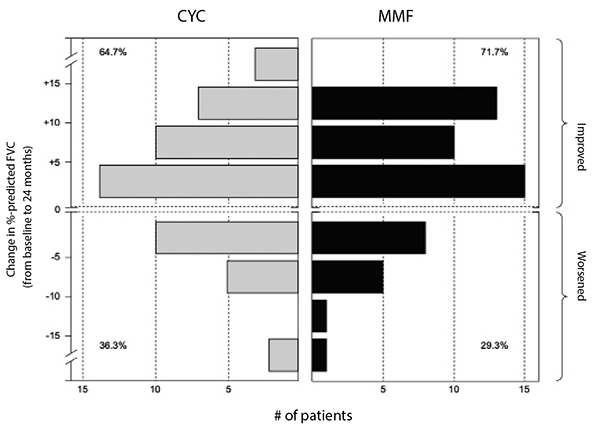
(click for larger image) Figure 2: Frequency distribution of changes from baseline to 24 months in FVC percent-predicted by treatment arm in SLS II.
Where these two drugs—mycophenolate mofetil and oral cyclophosphamide—diverged, however, was in their safety and tolerability profiles.6
Leukopenia (30 vs. four patients) and thrombocytopenia (four vs. zero patients) occurred significantly more frequently with cyclophosphamide than with mycophenolate. Frequent dose interruptions and titration setbacks occurred in the cyclophosphamide arm, but problems titrating mycophenolate were rare. Fewer patients on mycophenolate mofetil than on cyclophosphamide prematurely withdrew from the study drug (20 vs. 32) or met prespecified criteria for treatment failure (zero vs. two, defined as an absolute decline in FVC percent-predicted from baseline of 15%).
Taken together, the results of SLS II demonstrated mycophenolate mofetil is safer and better tolerated than cyclophosphamide.6 Although there was no difference in efficacy between these agents, it is plausible that advantages from mycophenolate were not detected due to initiation of alternative therapy in the second year of the study in the cyclophosphamide arm. For example, 12 of the 16 patients in the cyclophosphamide arm who prematurely stopped treatment but returned for the final study visit reported they had started an alternative therapy for their SSc-ILD (the most common agent used was mycophenolate).
Although there are no placebo-controlled trials for mycophenolate, an analysis comparing the mycophenolate arm of SLS II with the placebo arm of SLS I demonstrated that treatment with mycophenolate (compared with placebo) was associated with a significant improvement in the course of FVC percent-predicted, diffusing capacity, skin score and dyspnea over 24 months, even after adjusting for baseline disease severity.7 Moreover, patients assigned to mycophenolate suffered substantially fewer study drug withdrawals and serious adverse effects compared with patients assigned to placebo.7
Study #3
Based on the aforementioned studies, mycophenolate has emerged as a first-line treatment option for patients with SSc-ILD. However, as already noted, not all patients with SSc-ILD experience an improvement in their FVC percent-predicted when treated with mycophenolate, or with cyclophosphamide. In addition, the results of long-term follow-up analysis of patients who participated in SLS I and II revealed the leading cause of death in these patients was respiratory failure due to underlying ILD.8 Twelve years after the first patient was randomized in SLS I, 42% of participants had died (cyclophosphamide: 38; placebo: 28).8 Eight years after the first patient was randomized in SLS II, 21% of participants had died (cyclophosphamide: 16; mycophenolate: 14).8 Clearly, treatment with immunosuppression, at least for one to two years, is insufficient to halt the devastating clinical impact of ILD in certain patients with SSc.
Both cyclophosphamide and mycophenolate target predominantly inflammatory pathways implicated in SSc-ILD pathogenesis. Agents that specifically target fibrotic pathways may offer additional therapeutic benefit in this patient population, especially if combined with the well-tolerated immunosuppressant agent, mycophenolate. SLS III was specifically designed to test the hypothesis that the addition of anti-fibrotic therapy (i.e., pirfenidone) to a cytotoxic, immunosuppressant agent (i.e., mycophenolate) might lead to an improved treatment response compared with using an immunosuppressant agent alone.
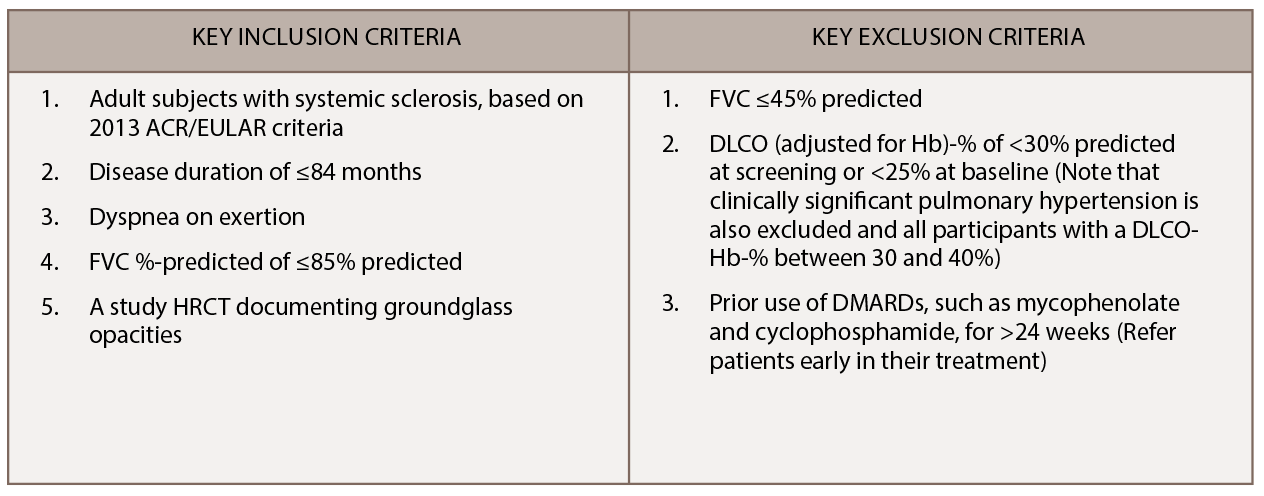
(click for larger image) Table 1: Key Inclusion & Exclusion Criteria for SLS III
For this study (currently open for enrollment and listed under identifier NCT03221257), patients with SSc-ILD are randomized to either treatment with mycophenolate alone or treatment with mycophenolate plus pirfenidone for 18 months. A prior open-label study demonstrated that pirfenidone is safe in patients with SSc-ILD. In that four-month study, patients were randomized in a 1:1 ratio to receive pirfenidone titrated over two or four weeks from a starting dose of 801 mg/day up to a maintenance dose of 2,403 mg/day. Mycophenolate mofetil (MMF) was combined with pirfenidone in 63.5% of patients with good tolerability, and the four-week dosage titration for pirfenidone was better tolerated than a two-week titration.9
SLS III is targeting SSc-ILD patients who live in North America and are treatment naive or early in their treatment course with MMF (i.e., treatment with MMF for 24 or fewer weeks prior to enrollment). This approach, combining both drugs up front, is designed to take advantage of the rapid onset of a treatment effect that has been observed when pirfenidone is used to treat idiopathic pulmonary fibrosis (not related to scleroderma). In contrast, we know from SLS II that the improvements in pulmonary function occur in a delayed manner when using MMF alone.
The hope: This early combination therapy will provide both a faster and synergistic effect of treatment on the course of FVC percent-predicted.
To date, no clinical trials in SSc-ILD patients have assessed the safety and efficacy of randomized treatment with an anti-fibrotic agent and an immunosuppressant agent. Although the SENSCIS trial (which compared nintedanib with placebo for SSc-ILD) allowed patients to be on background mycophenolate before and during the trial (or not), patients were not specifically randomized to combination, nor was the dosing of the mycophenolate standardized; the study showed statistically significant, but modest, change favoring nintedanib
Nintedanib in the annual rate of decline in FVC, with no improvement in skin score and patient-reported outcomes.10
We are recruiting additional sites in the U.S. and Canada for SLS III, with 16 active scleroderma centers currently participating in this trial. Learn more. SLS III is an investigator initiated trial funded by Genentech. The key entry criteria for SLS III appear in Table 1.
 Elizabeth R. Volkmann, MD, MS, is an assistant professor of medicine in the Division of Rheumatology at the University of California, Los Angeles (UCLA). She is the founder and co-director of the UCLA Connective Tissue Disease-Interstitial Lung Disease Program. She cares and advocates for patients with systemic sclerosis and autoimmune lung disease and is an active clinical and translational researcher in these areas.
Elizabeth R. Volkmann, MD, MS, is an assistant professor of medicine in the Division of Rheumatology at the University of California, Los Angeles (UCLA). She is the founder and co-director of the UCLA Connective Tissue Disease-Interstitial Lung Disease Program. She cares and advocates for patients with systemic sclerosis and autoimmune lung disease and is an active clinical and translational researcher in these areas.
 Michael D. Roth, MD, is a professor of pulmonary & critical care medicine and molecular toxicology at the David Geffen School of Medicine/UCLA and directs a combination of basic and translational research on understanding the lung and immunologic effects associated with marijuana and tobacco use, the development of immunologic therapies for cancer and more. He is also the vice chair for research compliance in the Department of Medicine. Dr. Roth has participated in all three of the Scleroderma Lung Studies and currently acts as the lead principal investigator/sponsor for Scleroderma Lung Study III.
Michael D. Roth, MD, is a professor of pulmonary & critical care medicine and molecular toxicology at the David Geffen School of Medicine/UCLA and directs a combination of basic and translational research on understanding the lung and immunologic effects associated with marijuana and tobacco use, the development of immunologic therapies for cancer and more. He is also the vice chair for research compliance in the Department of Medicine. Dr. Roth has participated in all three of the Scleroderma Lung Studies and currently acts as the lead principal investigator/sponsor for Scleroderma Lung Study III.
 Donald P. Tashkin, MD, is emeritus professor of medicine in the Division of Pulmonary and Critical Care Medicine at the David Geffen School of Medicine/UCLA. He served as director of the Clinical Pulmonary Function Laboratory at the UCLA Medical Center for approximately 30 years and as interim chief of his division. His research interests include the evaluation and treatment of SSc-ILD, as well as the pathophysiology, prevention and treatment of COPD, the pathophysiology and clinical pharmacology of asthma, the pulmonary effects of abuse of smoked substances (including marijuana, crack cocaine and tobacco) and of community air pollution.
Donald P. Tashkin, MD, is emeritus professor of medicine in the Division of Pulmonary and Critical Care Medicine at the David Geffen School of Medicine/UCLA. He served as director of the Clinical Pulmonary Function Laboratory at the UCLA Medical Center for approximately 30 years and as interim chief of his division. His research interests include the evaluation and treatment of SSc-ILD, as well as the pathophysiology, prevention and treatment of COPD, the pathophysiology and clinical pharmacology of asthma, the pulmonary effects of abuse of smoked substances (including marijuana, crack cocaine and tobacco) and of community air pollution.
 Cathie Spino, ScD, is a research professor and director of statistical analysis of biomedical and educational research (SABER) in the Department of Biostatistics. She collaborates with investigators on the design and analysis of multi-center clinical trials and provides infrastructure oversight. She joined the department in September 2007, after spending 10 years in the pharmaceutical industry as a statistician and statistical manager. She began her career in academia as an assistant professor in the Department of Biostatistics at the Harvard School of Public Health.
Cathie Spino, ScD, is a research professor and director of statistical analysis of biomedical and educational research (SABER) in the Department of Biostatistics. She collaborates with investigators on the design and analysis of multi-center clinical trials and provides infrastructure oversight. She joined the department in September 2007, after spending 10 years in the pharmaceutical industry as a statistician and statistical manager. She began her career in academia as an assistant professor in the Department of Biostatistics at the Harvard School of Public Health.
 Dinesh Khanna, MD, MSc, is the Frederick G.L. Huetwell Professor of Medicine and director, University of Michigan Scleroderma Program. Dr Khanna has published over 350 peer-reviewed articles and book chapters. His current research is focused on validating outcome measures, trial design and understanding the mechanism of action(s) of novel targets in the context of scleroderma clinical trials. He led the development of the ACR Combined Response Index in Systemic Sclerosis (ACR CRISS). Dr. Khanna is a recipient of the Henry Kunkel Young Investigator Award from the ACR.
Dinesh Khanna, MD, MSc, is the Frederick G.L. Huetwell Professor of Medicine and director, University of Michigan Scleroderma Program. Dr Khanna has published over 350 peer-reviewed articles and book chapters. His current research is focused on validating outcome measures, trial design and understanding the mechanism of action(s) of novel targets in the context of scleroderma clinical trials. He led the development of the ACR Combined Response Index in Systemic Sclerosis (ACR CRISS). Dr. Khanna is a recipient of the Henry Kunkel Young Investigator Award from the ACR.
Disclosures
This report describes an investigator-initiated and sponsored clinical trial supported in part by a funding contract from Genentech Inc. to the University of California, Los Angeles. Dr. Volkmann reports receiving consulting fees from Boehringer Ingelheim. Dr. Spino is a statistical consultant for Elcos.
Dr. Khanna is currently funded by the National Institutes of Health with grants from NIAMS and NIAID (as part of Clinical Autoimmunity Center of Excellence). He is the coordinating PI for ongoing and recently completed NIH and industry-funded trials of abatacept, brentuximab, pirfenidone, riociguat, tofacitinib and tocilizumab in scleroderma.
References
- Volkmann ER, Tashkin DP. Treatment of systemic sclerosis-related interstitial lung disease: A review of existing and emerging therapies. Ann Am Thorac Soc. 2016 Nov;13:2045–2056.
- Au K, Khanna D, Clements PJ, et al. Current concepts in disease-modifying therapy for systemic sclerosis-associated interstitial lung disease: Lessons from clinical trials. Curr Rheumatol Rep. 2009 Apr;11(2):111–119.
- Tashkin DP, Elashoff R, Clements PJ, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006 Jun 22;354(25):2655–2666.
- Kim HJ, Brown M, Elashoff F, et al. Quantitative texture-based assessment of one-year changes in fibrotic reticular patterns on HRCT in scleroderma lung disease treated with oral cyclophosphamide. Eur Radiol. 2011;21(12):2455–2465.
- Tashkin DP, Elashoff R, Clements PJ, et al. Effects of 1-year treatment with cyclophosphamide on outcomes at 2 years in scleroderma lung disease. Am J Respir Crit Care Med. 2007 Nov 15;176(10):1026–1034.
- Tashkin DP, Roth MD, Clements PJ, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease: Scleroderma lung study II (SLS-II), a double-blind, parallel group, randomised controlled trial. Lancet Respir Med. 2016 Sep;4(9):708–719.
- Volkmann ER, Tashkin DP, Li N, et al. Mycophenolate versus placebo for patients with systemic sclerosis-related interstitial lung disease. Arthritis Rheumatol. 2017 Jul;69(7):1451–1460.
- Volkmann ER, Tashkin DP, Sim M, et al. Short-term progression of interstitial lung disease in systemic sclerosis predicts long-term survival in two independent clinical trial cohorts. Ann Rheum Dis. 2019 Jan;78(1):122–130.
- Khanna D, Albera C, Fischer A, et al. An open-label, phase II study of the safety and tolerability of pirfenidone in patients with scleroderma-associated interstitial lung disease: The LOTUSS trial. J Rheumatol. 2016 Sep;43(9):1672–1679.
- Distler O, Brown KK, Distler JHW, et al. Design of a randomized, placebo-controlled clinical trial of nintedanib in patients with systemic sclerosis-associated interstitial lung disease (SENCSCIS). Clin Exp Rheumatol. 2017 Sep–Oct;35 Suppl 106(4):75–81.
