Classification of CTD-ILD Based on Histopathology
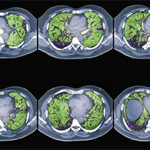
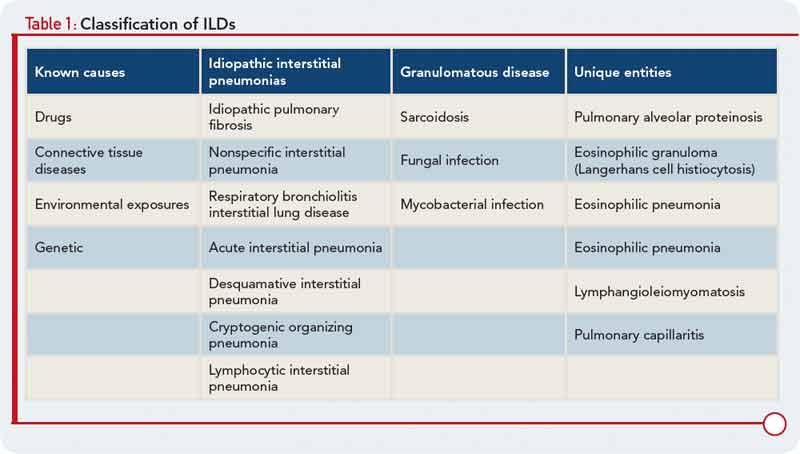
To characterize the histologic patterns in surgical lung biopsy (SLBx) specimens from patients with ILD, including those with CTD-ILD, pathologists use the classification scheme developed for the idiopathic interstitial pneumonias (IIPs).3 The IIPs are a group of seven ILDs of unknown cause, with potentially overlapping clinical and radiological features but different prognoses (see Table 1). Over the years, a challenging nomenclature and ever-changing classification scheme has created confusion around the IIPs. Also clouding understanding is the fact that, although the seven histologic patterns decisively distinguish one IIP from another, the patterns are not specific to “idiopathic” disease and may occur in ILD of any cause. For example, the fibrotic pattern of usual interstitial pneumonia (UIP pattern) has a number of potential causes, including chemotherapy-induced ILD, hypersensitivity pneumonitis, sarcoidosis, or any of the CTDs. The UIP pattern is the most common histological pattern in patients with IIP. However, among patients with CTD-ILD who undergo SLBx, the pattern of nonspecific interstitial pneumonia (NSIP pattern) is most frequently identified. It is unclear whether the NSIP pattern appears to predominate because of significant selection bias: relatively few patients with CTD-ILD undergo SLBx.