Case Study
A 26-year-old female was referred to the rheumatology service two years after the insidious onset of “walking funny” that progressed to significant proximal muscle weakness over a 10-month period. Early in the course of her illness, her creatine kinase (CK) was 4,000. Subsequent muscle biopsy revealed muscle fiber degeneration and regeneration in addition to numerous phagocytic cells. She was diagnosed with polymyositis but, despite treatment with high-dose corticosteroids, methotrexate (MTX), and monthly intravenous immunoglobulin (IVIG), she did not improve. At the time of presentation to rheumatology, her CK was 8,000, and she reported continued progression of muscle weakness and consequent frequent falls. Her family history was remarkable for a sister with the onset of similar symptoms at 30 years of age. Because of the lack of response to previous appropriate therapies and the family history, we suspected a hereditary myopathy. Her initial muscle biopsy results were consistent with the possibility of a hereditary myopathy, because some dystrophies do show inflammation. Repeat muscle biopsy was recommended for more extensive histopathologic evaluation. Upon immunohistochemical staining, the absence of dysferlin was discovered, consistent with a diagnosis of limb-girdle muscular dystrophy 2B.

Introduction
This case highlights the difficulty in diagnosing polymyositis and the need to think through the appropriate differential diagnosis and to use the appropriate diagnostic modalities when evaluating patients with suspected idiopathic inflammatory myopathy (IIM). Polymyositis (PM), dermatomyositis (DM), and inclusion body myositis (IBM) comprise the major IIM subsets in adults. A further categorization based on associated malignancy or other connective tissue disease features is useful in some circumstances, such as directing therapeutic interventions. Although the IIMs share the characteristics of immune-mediated attacks on skeletal muscle resulting in muscle weakness, they are in fact heterogeneous diseases with varied histopathological and clinical characteristics.1 Furthermore, these inflammatory myopathies can be confused with other myopathies that sometimes can have an inflammatory component, such as muscular dystrophies.
Differential Diagnosis
The differential diagnosis of a patient with suspected IIM is vast (see Table 1, p. 19) and highlights the importance of a meticulous history and physical, as well as appropriate and thorough diagnostic testing. The still-useful classification criteria of Bohan and Peter require four of the following features for a diagnosis of definite PM or three features plus a characteristic rash for a diagnosis of definite DM:2,3
- Symmetric, subacute muscle weakness;
- Abnormal muscle biopsy: fiber size variation, necrosis, regeneration, atrophy, and inflammation;
- Muscle enzyme elevation: CK, aldolase, alanine transaminase (ALT), aspartate aminotransferase (AST), and lactic dehydrogenase (LDH);
- Electromyogram abnormality: triad of short, small, polyphasic motor units; insertional irritability, positive sharp waves, fibrillations; and bizarre, high-frequency repetitive discharges; and
- Typical DM skin abnormalities: Gottron’s papules (see Figure 2, below) or heliotrope rash.
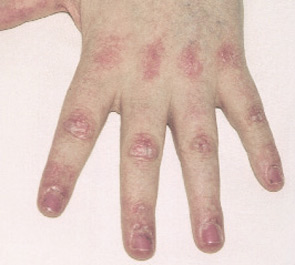
Exclusion criteria include evidence of metabolic, infectious, post-traumatic, and neuromuscular disorders.2,3 Although widely cited, Bohan and Peter’s criteria were developed prior to the discovery of myositis-specific autoantibodies (MSAs) and have been criticized for their inability to differentiate patients with PM, IBM, the toxic myopathies, and some genetic and metabolic myopathies.1