Developments in CPPD Epidemiology
In most patients, CPPD appears to represent a systemic articular and soft tissue pathophysiology disorder in aging, a paradigm reinforced by the results of the unique Genetics of Osteoarthritis and Lifestyle study cohort (GOAL).1,2 In GOAL, radiographic evidence of CPPD at one joint was frequently associated with CPPD at distant joints, whether osteoarthritis (OA) was present or not.2 One unexpected nuance of GOAL was that knee OA, but not hip OA, was associated with radiographic evidence of CPPD at distant joints.2
True prevalence of CPPD as a pathologic finding, let alone symptomatic arthritis, has not yet been reliably measured. This is due to limits of both survey methods and plain radiographic detection, and is highlighted by findings of plain radiographs of knees, hands, and pelvis in roughly equal-sized, large groups with clinically severe hip OA, clinically severe knee OA (defined as radiographic OA, and needing joint arthroplasty), and without knee or hip OA in GOAL.1,2 The knee is the most common site of radiographic chondrocalcinosis, but 42% of the chondrocalcinosis cases in GOAL had no knee involvement.1 This underscores the need, in clinical practice, to do thorough, joint-specific screening for CPPD, where appropriate, rather than knee radiographs to rule in or rule out CPPD at distant joints.
Environmental and genetic factors likely underlie differences in development and clinical phenotype of CPPD in populations. In this context, a markedly decreased prevalence of CPPD in a random sample of Chinese patients aged 60 years and older in Beijing, China, compared to a control population of white patients in Framingham, Mass., is surprising, given the excess of knee OA in the Chinese group of this study cohort.3 Environmental influences on CPPD could include dietary mineral content and other factors regulating body iron, calcium, phosphate, and magnesium stores, and parathyroid function and catalytic activity of enzymes that regulate inorganic pyrophosphate (PPi) formation and hydrolysis.3 Acquired and genetic factors likely include effects of dysregulated mitochondrial function on adenosine triphosphate (ATP) and PPi metabolism in aging and OA.4,5
Recent epidemiologic studies suggest a growing CPPD prevalence in developed countries, not simply due to increased longevity of the population, but to iatrogenic factors as well. Examples include hypomagnesemia, promoted by increased prescription of loop and thiazide diuretics, proton pump inhibitors, and calcineurin inhibitors (cyclosporine, tacrolimus), and by iatrogenic short bowel syndrome.6
Advances in CPPD Pathogenesis and Molecular Genetics
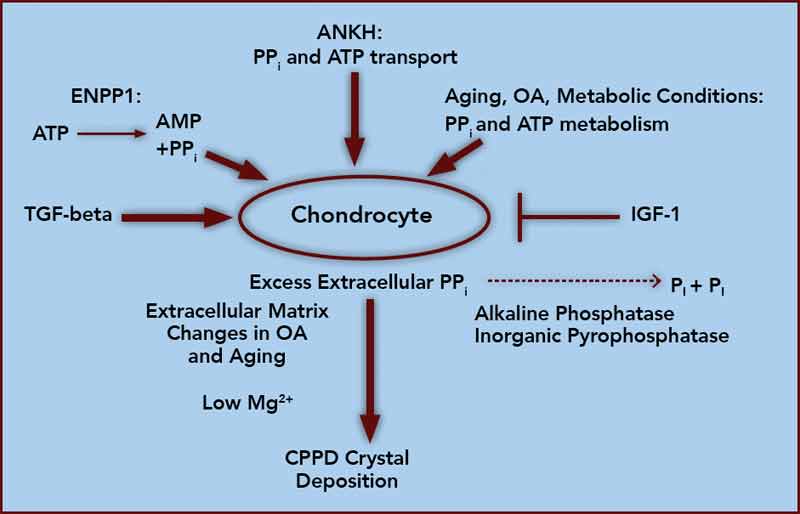
At the core of crystal deposition in idiopathic and other forms of CPPD is dysregulated PPi metabolism, accompanied by altered chondrocyte differentiation and function (see Figure 1).7,8 Articular cartilage, unlike growth-plate cartilage, is specialized to avoid the process of matrix calcification. Relatively copious output of PPi by chondrocytes physiologically functions to limit basic calcium phosphate crystal growth. This protective mechanism becomes deleterious by excessive PPi output in CPPD.7,8 Factors responsible for upregulating PPi generation in the aging joint and in OA are likely heterogeneous, but most function through increasing articular ectonucleotide pyrophosphatase phosphodiesterase (ENPP1) and the multiple-pass, transmembrane protein ankylosis protein homolog human gene (ANKH).7,8
ENPP1 generates intracellular and extracellular PPi via hydrolysis of ATP and other nucleoside triphosphates, and articular ecto–nucleotide pyrophosphatase/phosphodiesterase (ecto-NPP) activity is substantially elevated in both CPPD and OA.8 Intracellular PPi and ATP are transported to the extracellular space by processes involving ANKH, whose expression rises in OA chondrocytes.7,8
Players in aging- and OA-associated alterations in PPi metabolism also include dysregulated chondrocyte responses to growth factors (See Figure 1).8-10 Transforming growth factor-β (TGF-β) increases, insulin-like growth factor-1 (IGF-1) decreases chondrocyte extracellular PPi, and calcium (Ca2+) potentiates the effects of TGF-β on PPi.8-10 Certain chondrocyte responses to TGF-β—including PPi generation—increase in aging cartilage, possibly via known changes in TGF-β receptor subunit expression. Moreover, IGF-1 resistance increases in aging and OA cartilage, due to a variety of factors.8-10
Ambient cartilage magnesium (Mg2+) concentration, and multiple chondrocyte extracellular matrix components, including collagens II and I, proteoglycans, and osteopontin, also modulate crystal deposition in CPPD, and Mg2+ helps determine whether more inflammatory monoclinic or less inflammatory triclinic CPPD crystals are formed.8,11 We now recognize that the inflammatory responses to crystals in CPPD, as in gout, are mediated by NOD-like receptor, pyrin domain containing 3 (NLRP3) inflammasome activation and consequent caspase-1 activation, and interleukin 1-β (IL-1β) maturation and secretion.12
Exciting molecular genetic advances in CPPD have been spurred by identification of linkage of familial, early-onset CPPD with multiple mutations in ANKH.7,8 Moreover, homozygosity for -4 G to A substitution in the ANKH 5’-untranslated region, which promotes increased ANKH mRNA levels, was present in approximately 4% of British subjects who had carried a prior diagnosis of idiopathic chondrocalcinosis of aging.13 Increased ANKH promotes cartilage matrix catabolism by chondrocytes.8 As such, ANKH mediates development and anatomic progression of both early-onset familial CPPD and late-onset disease.
New Developments in CPPD Clinical Features and Outcomes
A recent consensus process by the European League Against Rheumatism (EULAR) reclassified CPPD, and suggested new terminology for clinical forms of CPPD arthritis.14 EULAR’s proposed abandonment of the “pseudogout” terminology for acute CPPD arthritis ultimately may be counterproductive for disease recognition outside rheumatology. Other limits of the EULAR classification, in my opinion, include lack of precise attention to systemic degenerative arthropathy primarily driven by CPPD in a subset of patients, rather than secondarily associated with the primary process of OA in the majority. In this context, chronic degenerative arthropathy in primary CPPD commonly affects certain joints that are typically spared by primary OA, such as the metacarpophalangeal and wrist joints.15
CPPD is very often clinically silent, but is a biomarker of aging, connective-tissue degeneration, and low-grade inflammation.15 Because CPPD is so strongly linked with OA, and can manifest as a primary cause of inflammation and cartilage-matrix catabolism, the impression has been that CPPD drives inflammatory symptoms and progression in OA. However, there is no clear correlation between the extent of calcification and progression of CPPD. The Boston Osteoarthritis of the Knee Study and the Health, Aging, and Body Composition Study did not link CPPD with elevated risk of cartilage loss in OA.16 Other work revealed no significant differences with or without CPPD in activities of daily living, or age at total knee arthroplasty, for degenerative arthritis.15 Moreover, the association between OA and CPPD may be linked more to presence of osteophytes than to joint-space narrowing.15
CPPD is likely a biomarker of cartilage aging in most circumstances, and, in OA, CPPD may be a biomarker of a dysregulated, but functionally significant, cartilage repair response, and of noxious effects of excess extracellular PPi independent of crystal deposition.8 However, this does not mean that CPPD is simply an epiphenomenon. An illustrative parallel is the dual implication, as biomarker and mediator of tissue complications, of basic calcium phosphate crystal deposition in OA, tendinopathies such as shoulder-rotator-cuff disease, and in arteries in atherosclerosis and diabetic and end-stage renal disease arteriopathies.
Emerging Trends in CPPD Diagnosis
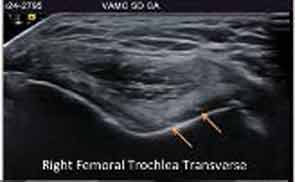
Major advances in CPPD diagnostics include formal recognition by EULAR of the utility of high-resolution ultrasound, now a standard tool for gout diagnosis.14 Ultrasound is convenient, inexpensive, does not involve radiation, picks up even subtle joint inflammation, correlates with positive results by synovial fluid crystal analysis, and can differentiate between gout and CPPD in most circumstances.7 In addition, ultrasound can detect CPPD even in the absence of chondrocalcinosis by plain radiography (see Figure 2).17
Preliminary criteria proposed for CPPD by ultrasound have been published.17 However, false positives and negatives can occur. The approach is best for CPPD detection in fibrocartilages and in middle zones of articular hyaline cartilages. Diagnostic value for CPPD disease in studies of plantar fascia and tendons such as the Achilles remains unclear. Whether dual-energy computed tomography (CT) can be usefully applied to CPPD diagnosis is not resolved, and magnetic resonance imaging is not yet optimized for diagnosis of CPPD.
Rheumatologists increasingly recognize that CPPD is a tremendous disease mimic, going well beyond gout, OA, RA, and septic arthritis to include polymyalgia rheumatica and meningitis.14,15 Given that CPPD is so common, accurate exclusion of entities other than CPPD is accepted clinical practice in acute and chronic arthropathies in which CPPD is present. In particular, since infectious arthritis can coexist with CPPD and gout, imaging approaches do not render synovial fluid analysis obsolete in the diagnosis of acute inflammatory arthritis.

Current and Emerging Treatment for CPPD Clinical Manifestations
EULAR-expert, opinion-based recommendations emphasize that treatment of acute CPPD arthritis is similar to that for acute gout, but with a starkly contrasting lack of randomized, controlled clinical trials for CPPD.18 There are many available modalities (see Table 2). For example, intraarticular steroids are useful, particularly in elderly subjects with multiple comorbidities.15,18 Likely benefits exist for prophylaxis of acute CPPD with low-dose colchicine, and practitioners need to be aware of risks for precipitating acute CPPD arthritis in the postoperative state (including postparathroidectomy, and in affected knee joints after arthroplasty) and after injecting intraarticular hyaluronan, or prescribing bisphosphonates or granulocyte colony–stimulating factor (G-CSF).15
For recurrent acute arthritis and refractory chronic arthritis, there are possible benefits, described in small, inadequately controlled studies of hydroxychloroquine (presumably by decreasing phagolysosomal-mediated crystal-induced NLRP3 inflammasome activation) and methotrexate.15,19 Conversely, rebound arthritis after tapering systemic corticosteroids could be an issue mediated by the known ability of corticosteroids to induce NLRP3 production in macrophages. There is no clear benefit of anti–tumor necrosis factor (TNF) therapy in CPPD. However, interleukin 1 antagonism clearly merits further investigation for refractory CPPD arthritis, as in gout.20
It is not yet clear whether oral magnesium supplementation will have benefits in inhibiting crystal deposition in established CPPD.15 For example, promotion of crystal dissolution by recombinant alkaline phosphatase risks flares of acute arthritis by crystal shedding from cartilage.15 ENPP1 activity, and ANKH transport function (which is nonselectively inhibited by probenecid) represent compelling targets for future molecular therapeutics to suppress crystal deposition in severe, symptomatic CPPD.15

Conclusion
Writer Hugh Prather described life as “a mixture of unsolved problems, ambiguous victories, and vague defeats with very few moments of clear peace.” So it is metaphorically in rheumatology, at present, with CPPD. The last decade has seen substantial advances in the disorder, but huge hurdles remain (see Table 3). A good starting point would be a multicenter, randomized, controlled clinical trial of symptomatic disease subsets driven by CPPD.
Acknowledgment
This work was supported by the Veteran’s Affairs Research Service and the National Institutes of Health grant PAG07996. Dr. Terkeltaub is a consultant to Takeda Pharmaceutical Company Ltd., Pfizer Inc., Regeneron Pharmaceuticals Inc., and Novartis Pharmaceuticals.
Dr. Terkeltaub is chief of the rheumatology section at the Veterans Affairs Medical Center in San Diego, and a professor of medicine at the University of California, San Diego School of Medicine.
References
- Abhishek A, Doherty S, Maciewicz R, Muir K, Zhang W, Doherty M. Chondrocalcinosis is common in the absence of knee involvement. Arthritis Res Ther. 2012;14:R205.
- Abhishek A, Doherty S, Maciewicz R, Muir K, Zhang W, Doherty M. Evidence of a systemic predisposition to chondrocalcinosis and association between chondrocalcinosis and osteoarthritis at distant joints: A cross-sectional study. Arthritis Care Res (Hoboken). 2013;65:1052-1058.
- Zhang Y, Terkeltaub R, Nevitt M, et al. Lower prevalence of chondrocalcinosis in Chinese subjects in Beijing than in white subjects in the United States: The Beijing osteoarthritis study. Arthritis Rheum. 2006;54:3508-3512.
- Johnson K, Jung A, Murphy A, Andreyev A, Dykens J, Terkeltaub R. Mitochondrial oxidative phosphorylation is a downstream regulator of nitric oxide effects on chondrocyte matrix synthesis and mineralization. Arthritis Rheum. 2000;43:1560-1570.
- Johnson K, Svensson CI, Etten DV, et al. Mediation of spontaneous knee osteoarthritis by progressive chondrocyte ATP depletion in Hartley guinea pigs. Arthritis Rheum. 2004;50:1216-1225.
- Rho YH, Zhu Y, Zhang Y, Reginato AM, Choi HK. Risk factors for pseudogout in the general population. Rheumatology (Oxford). 2012;51:2070-2074.
- Abhishek A, Doherty M. Pathophysiology of articular chondrocalcinosis—role of ANKH. Nat Rev Rheumatol. 2011;7:96-104.
- Terkeltaub R, Pritzker KPH. Pathogenesis and molecular genetics of calcium pyrophosphate dihydrate crystal deposition disease. In: Terkeltaub R, ed. Gout and Other Crystal Deposition Arthropathies. Philadelphia, Penn: Elsevier; 2011:240-248.
- Costello JC, Rosenthal AK, Kurup IV, Masuda I, Medhora M, Ryan LM. Parallel regulation of extracellular ATP and inorganic pyrophosphate: Roles of growth factors, transduction modulators, and ANK. Connect Tissue Res. 2011;52:139-146.
- Johnson K, Farley D, Hu SI, Terkeltaub R. One of two chondrocyte-expressed isoforms of cartilage intermediate-layer protein functions as an insulin-like growth factor 1 antagonist. Arthritis Rheum. 2003;48:1302-1314.
- Jubeck B, Muth E, Gohr CM, Rosenthal AK. Type II collagen levels correlate with mineralization by articular cartilage vesicles. Arthritis Rheum. 2009;60:2741-2746.
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237-241.
- Zhang Y, Johnson K, Russell RG, et al. Association of sporadic chondrocalcinosis with a -4-basepair G-to-A transition in the 5’-untranslated region of ANKH that promotes enhanced expression of ANKH protein and excess generation of extracellular inorganic pyrophosphate. Arthritis Rheum. 2005;52:1110-1117.
- Zhang W, Doherty M, Bardin T, et al. European League Against Rheumatism recommendations for calcium pyrophosphate deposition. Part I: Terminology and diagnosis. Ann Rheum Dis. 2011;70:563-570.
- Guerne PA, Terkeltaub R. Calcium pyrophosphate dihydrate crystal deposition: Epidemiology, clinical features, diagnosis, and treatment. In: Terkeltaub R, ed. Gout and Other Crystal Deposition Arthropathies. Philadelphia, Penn: Elsevier; 2011:249-265.
- Neogi T, Nevitt M, Niu J, et al. Lack of association between chondrocalcinosis and increased risk of cartilage loss in knees with osteoarthritis: Results of two prospective longitudinal magnetic resonance imaging studies. Arthritis Rheum. 2006;54:1822-1828.
- Frediani B, Filippou G, Falsetti P, et al. Diagnosis of calcium pyrophosphate dihydrate crystal deposition disease: Ultrasonographic criteria proposed. Ann Rheum Dis. 2005;64:638-640.
- Zhang W, Doherty M, Pascual E, et al. EULAR recommendations for calcium pyrophosphate deposition. Part II: Management. Ann Rheum Dis. 2011;70:571-575.
- Chollet-Janin A, Finckh A, Dudler J, Guerne PA. Methotrexate as an alternative therapy for chronic calcium pyrophosphate deposition disease: An exploratory analysis. Arthritis Rheum. 2007;56:688-692.
- Announ N, Palmer G, Guerne PA, Gabay C. Anakinra is a possible alternative in the treatment and prevention of acute attacks of pseudogout in end-stage renal failure. Joint Bone Spine. 2009;76:424-426.